Интерстициальные заболевания легких (ИЗЛ) занимают до 10–15 % в структуре всех болезней легких. К ним относят более 130 заболеваний известной и неизвестной этиологии. Одним из наиболее распространенных и прогностически неблагоприятных заболеваний из группы ИЗЛ является идиопатический фиброзирующий альвеолит (ИФА). ИФА — это заболевание легких неясной природы с морфологической картиной обычной интерстициальной пневмонии, характеризующееся нарастающей легочной недостаточностью вследствие развития преимущественно в интерстициальной ткани легких небактериального воспаления, ведущего к прогрессирующему интерстициальному фиброзу и в конечном итоге к смерти больного.
 Впервые заболевание было описано L. Hamman и A. Rich в 1935 г. на примере 4 больных с быстропрогрессирующей дыхательной недостаточностью, умерших в течение 6 мес. с момента заболевания. На вскрытии был обнаружен выраженный распространенный фиброз легких, и заболевание получило название «острый диффузный интерстициальный фиброз легких». Довольно долго синдромом Хаммена — Рича называли и заболевания с хроническим течением, но в последние годы этот синдром относят лишь к одной из форм ИФА — быстропрогрессирующей или острой интерстициальной пневмонии. Первым описал заболевание немец G. Rindfleisch (1897). Он предложил для него термин «кистозный цирроз легких». Термин «фиброзирующий альвеолит», предложенный J. Scadding позже, в 1964 г., отражает основные ключевые признаки заболевания — воспаление и фиброз. Синонимами «ИФА» являются «идиопатический легочный фиброз» и «криптогенный фиброзирующий альвеолит», а также «обычная интерстициальная пневмония», что отражает наиболее частый морфологический субстрат заболевания.
Впервые заболевание было описано L. Hamman и A. Rich в 1935 г. на примере 4 больных с быстропрогрессирующей дыхательной недостаточностью, умерших в течение 6 мес. с момента заболевания. На вскрытии был обнаружен выраженный распространенный фиброз легких, и заболевание получило название «острый диффузный интерстициальный фиброз легких». Довольно долго синдромом Хаммена — Рича называли и заболевания с хроническим течением, но в последние годы этот синдром относят лишь к одной из форм ИФА — быстропрогрессирующей или острой интерстициальной пневмонии. Первым описал заболевание немец G. Rindfleisch (1897). Он предложил для него термин «кистозный цирроз легких». Термин «фиброзирующий альвеолит», предложенный J. Scadding позже, в 1964 г., отражает основные ключевые признаки заболевания — воспаление и фиброз. Синонимами «ИФА» являются «идиопатический легочный фиброз» и «криптогенный фиброзирующий альвеолит», а также «обычная интерстициальная пневмония», что отражает наиболее частый морфологический субстрат заболевания.
Данные о распространенности ИФА и летальности вследствие него значительно варьируют. Это связано в первую очередь с существовавшей терминологической неопределенностью. Распространенность ИФА составляет 20,2 случая на 100 тыс. среди мужчин и 13,2 — среди женщин. Заболеваемость ИФА достигает 11,3 случая в год на 100 тыс. среди мужчин и 7,1 — среди женщин, увеличиваясь с возрастом. Примерно 2/3 пациентов с ИФА — старше 60 лет.
Смертность от ИФА больше в старшей возрастной группе и составляет в среднем 3,0 на 100 тыс. населения, медиана выживаемости колеблется от 2,3 года до 5 лет.
Данные различия в показателях распространенности и летальности ИФА очень трудно анализировать, так как в основе лежат различные подходы к кодированию заболевания в национальных регистрах, различия в экологических условиях, уровне диагностики заболевания и других факторах. Кроме того, основные исследования ИФА, как правило, проводят в специализированных пульмонологических центрах, где наблюдают более молодых и более тяжелых больных, что значительно искажает тенденции распространения и развития заболевания в мире.
Несмотря на наличие в названии заболевания термина «идиопатический» или «криптогенный», в настоящее время ученые ищут причины возникновения ИФА. Заболевание рассматривается как процесс, протекающий в несколько этапов: 1) первичное повреждение эпителиальных и/или эндотелиальных клеток легочной паренхимы с развитием воспаления; 2) регенерация структуры поврежденной ткани с накоплением мезенхимальных клеток и избыточным развитием экстрацеллюлярного матрикса/фиброза.
Существовавшие многие годы теории вирусного, аутоиммунного, наследственного и полиэтиологического происхождения ИФА пока не получили доказательств. Сегодня приходится говорить о каком-то неизвестном причинном факторе, который запускает каскад стереотипных реакций легочной ткани, рассматриваемых как воспаление. Оно проявляется скоплением и активацией клеток-эффекторов (нейтрофилы, макрофаги, лимфоциты) с формированием интерстициального и внутриальвеолярного отека, дезорганизацией структурной основы альвеол, интерстиция и терминальных бронхиол, изменением количественного и качественного состава сурфактанта. Эти деструктивные изменения происходят одновременно с патологически усиленным репаративным процессом в виде пролиферации фибробластов и отложения коллагена, что в конечном итоге приводит к формированию легочного фиброза с вытекающими отсюда вентиляционными, гемодинамическими и системными эффектами. При этом процесс фиброзирования настолько интенсивен, что порой не соответствует повреждению, его вызвавшему. Это дает основание предполагать первичную роль фиброзирования в патогенезе ИФА без существенного влияния на него воспалительного процесса. По мере прогрессирования фиброза и легочной недостаточности развивается хроническое легочное сердце.
Также считается, что существует генетическая предрасположенность к развитию ИФА в ответ на неспецифическое повреждение эпителия. В пользу данной гипотезы говорит наличие семейных форм заболевания. Изучается генетическая предрасположенность к развитию диффузных заболеваний легких, в основе которой лежит наследственный полиморфизм генов, кодирующих протеины, участвующие в процессинге и презентации антигенов к T-лимфоцитам.
Гистологическая картина при ИФА неоднородна. В одной из классификаций выделяют семь морфологических типов ИФА:
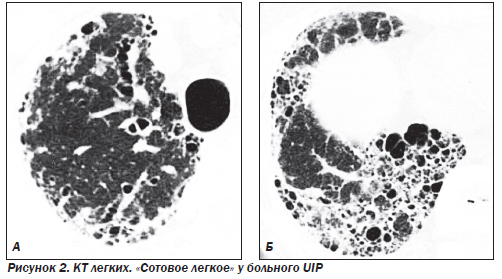
Наиболее частым гистологическим паттерном является обычная интерстициальная пневмония — UIP (синонимы: муральная форма или смешанный фиброзно-воспалительный вариант ИФА), которая составляет около 90 % всех форм ИФА. На ранних стадиях заболевания морфологическая картина характеризуется отеком и интенсивной инфильтрацией стенок альвеол лимфоцитами, моноцитами, плазматическими клетками и эозинофилами. Наряду с воспалением присутствуют признаки фиброза: фокусы фибробластов, активно синтезирующих коллаген, миофибробласты, сократительная способность которых может играть определенную роль в ремоделировании паренхимы легких. Часто, особенно на фоне диссеминированных заболеваний соединительной ткани (ДЗСТ), присутствуют признаки фокальной гиперплазии лимфоидной ткани. На более поздних стадиях заболевания происходит замещение нормальной структуры паренхимы грубой соединительной тканью, в которую замурованы кистозно расширенные воздухоносные пространства, выстланные изнутри гиперплазированным бронхиолярным или кубовидным альвеолярным эпителием. Альвеолоциты 1-го типа замещаются альвеолоцитами 2-го типа, иногда присутствуют признаки метаплазии. В полях фиброза могут присутствовать клетки воспаления, однако их количество не такое выраженное, как на ранних стадиях. Также в полях фиброза часто находят реактивную мышечную гиперплазию. Внутри измененных альвеол могут обнаруживаться муцин, белковый детрит, кристаллы холестерина, макрофаги и другие клетки. Стенки сосудов значительно утолщены в области фиброзных полей и могут быть нормальными в непораженной ткани легких. Макроскопические изменения легких на поздних стадиях характеризуются уплотнением и сморщиванием ткани легких и формированием «сотового легкого» (на этом основании одним из первых названий болезни было «кистозный цирроз легких»). Фиброз особенно выражен в субплевральной области и напоминает ленту шириной несколько сантиметров. Поражение плевры в отличие от асбестоза встречается редко. Прогноз неблагоприятный, летальность в течение пяти лет превышает 60 %.
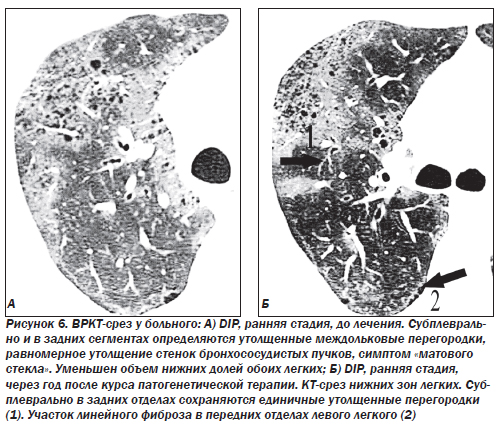
На долю десквамативной интерстициальной пневмонии (DIP) приходится около 5 % ИФА. Отличием данной формы является содержание большого количества клеток в просвете альвеол. Считалось, что основными типами клеток являются десквамированные альвеолоциты, что и легло в основу названия данного паттерна. Однако внутри альвеол в основном обнаруживаются макрофаги. Альвеолы выстланы гиперплазированными альвеолоцитами 2-го типа. Альвеолярные септы инфильтрированы лимфоцитами, иногда эозинофилами, может наблюдаться небольшое повышение содержания мезенхимальных клеток, однако фиброз, как правило, выражен незначительно. До сих пор не ясно, является ли десквамативная пневмония самостоятельным заболеванием или ранним проявлением обычной интерстициальной пневмонии. Настоящая форма DIP отличается чувствительностью к кортикостероидам и хорошим прогнозом, однако иногда болезнь прогрессирует до стадии «сотового легкого». Летальность при десквамативной пневмонии не превышает 25 %.
Острая интерстициальная пневмония (AIP) была впервые описана L. Hamman и A. Rich в 1935 г., однако в самостоятельную форму была выделена в лишь в 1986 г. В настоящее время только данная форма ИФА может обозначаться как синдром Хаммена — Рича. Встречается редко (примерно в 1 % случаев). Картина морфологических изменений при АIP идентична картине диффузного альвеолярного повреждения, наблюдающегося при респираторном дистресс-синдроме взрослых. В раннюю фазу отмечаются интерстициальный отек паренхимы и формирование гиалиновых мембран, на поздних этапах (после второй недели от начала заболевания) — признаки интраальвеолярной и/или интерстициальной организации, обычно ассоциированные с пролиферацией альвеолоцитов 2-го типа. Часто выявляются тромбы в мелких артериях. Прогноз неблагоприятный, летальность может достигать 90 %.
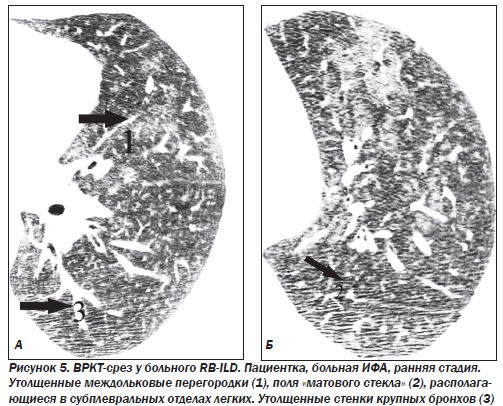
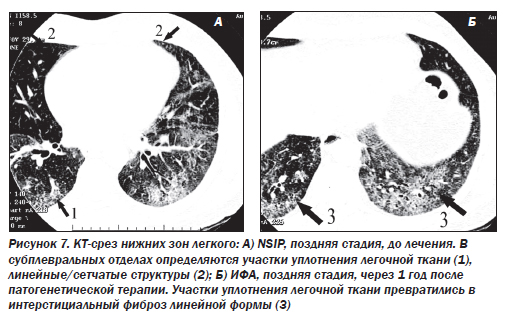
В 1994 г. был описан новый подтип ИФА — неспецифическая интерстициальная пневмония/фиброз (NSIP) (неклассифицируемая пневмония). Морфологическая картина отлична от всех известных вариантов. Эта форма выявляется у 5 % больных. Гистологически это целлюлярная интерстициальная пневмония с однородной картиной, характеризующейся лимфоплазмоцитарной инфильтрацией в пределах альвеолярных перегородок. В некоторых случаях наблюдается аккумуляция макрофагов в просвете альвеол, однако по сравнению с DIP при неспецифической пневмонии данный феномен имеет «пятнистый», негомогенный тип распределения, преобладает интерстициальное воспаление. Эта форма ИФА является самой благоприятной и характеризуется подострым течением. Более чем в 80 % случаев при NSIP наблюдается регрессирование или стабилизация воспалительного процесса, летальность не превышает 11–17 %.
Лимфоидная интерстициальная пневмония (LIP) встречается редко, обычно у женщин, чаще старше 40 лет. Характерно медленное развитие заболевания, одышка и кашель постепенно нарастают в течение 3–4 лет и более. Характерны лихорадка, боль в груди, артралгии, похудание. В легких выслушиваются трескучие хрипы. Могут наблюдаться изменения в крови — анемия и гипергаммаглобулинемия.
Заболевание поддается терапии глюкокортикоидами (ГК) и имеет благоприятный прогноз, однако примерно у 1/3 пациентов формируется диффузный интерстициальный фиброз.
В 2001 г. были описаны новые формы ИФА — криптогенная организующая пневмония и респираторный бронхиолит (COP и RB-ILD соответственно).
COP, криптогенная организующая пневмония, характеризуется вовлечением в патологический процесс дистальных воздушных пространств — альвеолярных ходов и альвеол в сочетании с полипоидным бронхиолитом или без него.
COP одинаково часто встречается у мужчин и женщин, чаще всего — у лиц старше 55 лет. Симптомы заболевания обычно сохраняются менее 3 мес. Характерно гриппоподобное начало болезни с кашля, лихорадки, миалгий, недомогания. Кашель чаще продуктивный с выделением прозрачной бесцветной мокроты. В легких выслушиваются локализованные или распространенные трескучие хрипы. Форма ногтевых фаланг не изменяется.
Симптомы обычно расцениваются как проявление инфекции нижних дыхательных путей, в связи с чем многим больным безуспешно проводится антибиотикотерапия.
При лабораторном исследовании крови выявляют повышение содержания С-реактивного белка, нейтрофилов, увеличение СОЭ. При исследовании ФВД определяются умеренные рестриктивные расстройства, снижение диффузионной способности легких, возможна небольшая артериальная гипоксемия.
При назначении глюкокортикоидов у большинства больных наступает полное выздоровление. Однако в период от 1 до 3 мес. после прекращения ГК-терапии или уменьшения дозы ниже 15 мг в сутки часто наблюдаются рецидивы заболевания. В связи с этим продолжительность ГК-терапии должна быть не менее 6 мес.
RB-ILD характеризуется поражением респираторных бронхиол с наличием пигментированных макрофагов в их стенках, сочетающимся с интерстициальным заболеванием легких.
RB-ILD — болезнь курильщиков со стажем более 30 пачко-лет. У большинства пациентов симптомы заболевания выражены незначительно, но у части больных может развиваться тяжелая одышка и гипоксемия. Заболевание начинается постепенно: появляется или усиливается кашель, начинается одышка. При физикальном обследовании патологические изменения в легких часто не определяются, у части больных могут выслушиваться трескучие хрипы.
Отличительной чертой при исследовании ФВД является наличие не только рестриктивных, но и обструктивных нарушений легочной вентиляции с увеличением остаточного объема легких. Отмечается также умеренное снижение диффузионной способности легких.
Клиническое течение и прогноз RB-ILD чаще благоприятны. Прекращение курения, как правило, обусловливает уменьшение выраженности одышки. Эффективна терапия ГК.
Клиническая картина ИФА не имеет патогномоничных признаков. Заболевание чаще всего встречается у пациентов в возрасте от 40 до 70 лет (чаще у мужчин). Основными жалобами являются одышка и сухой кашель. По мере развития отмечается нарастание одышки, вплоть до полной инвалидизации больного: из-за одышки больной не способен сказать даже несколько слов, не может сам себя обслуживать. Начало болезни чаще всего незаметное, но иногда ИФА может начинаться острыми симптомами, что предполагает роль вирусной инфекции в патогенезе заболевания. Из-за медленного прогрессирования заболевания пациенты успевают адаптироваться к одышке, постепенно снижая свою активность и переходя к более пассивному образу жизни. Большинство пациентов на момент обследования уже больны от 1 до 3 лет. Иногда отмечается кашель с мокротой, причем данный признак считается неблагоприятным. Лихорадка не характерна для ИФА. Другими симптомами могут быть общая слабость, артралгии, миалгии, снижение массы тела, изменение ногтевых фаланг (в виде «барабанных палочек»). Артриты и артралгии чаще встречаются у женщин, чем у мужчин. Симптом «барабанных палочек», наоборот, преобладает у мужчин.
Характерным аускультативным феноменом при ИФА является крепитация («треск целлофана»). Хрипы наиболее часто выслушиваются в заднебазальных отделах, хотя в 20 % всех случаев крепитацию можно прослушать и в верхних отделах. По сравнению с крепитацией при других заболеваниях (пневмония, бронхоэктазы, застойные процессы в легких) крепитация при ИФА более нежная — менее громкая и более высокая по частоте, выслушивается в конечно-инспираторную фазу. Иногда можно выслушать и экспираторную крепитацию (чаще во вторую треть выдоха), что может быть признаком прогрессирования заболевания. Сухие хрипы могут быть слышны у 5–10 % больных и обычно появляются при сопутствующем бронхите. У 50 % всех пациентов отмечается тахипноэ. По мере прогрессирования заболевания появляются дыхательная недостаточность и легочное сердце: диффузный серо-пепельный цианоз, усиление 2-го тона над легочной артерией, тахикардия, набухание шейных вен, периферические отеки. Выживаемость пациентов с момента постановки диагноза — в среднем 3–5 лет. Прогноз лучше у женщин, у более молодых пациентов, при анамнезе заболевания менее 1 года и у больных ИФА на фоне ДЗСТ. Наиболее частой причиной смерти больных является дыхательная недостаточность. На фоне ИФА возможно развитие любой формы рака легкого: наиболее частыми гистологическими вариантами являются плоскоклеточный рак и аденокарцинома, описано развитие бронхоальвеолярного и гигантоклеточного рака. Вообще риск заболевания раком легких у больных ИФА выше в 14 раз, чем в общей популяции того же возраста, пола и длительности курения.
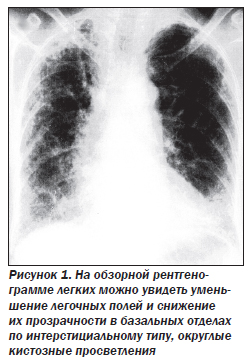
Для диагностики ИФА рентгенография грудной клетки является одним из важнейших диагностических методов. Наиболее частый признак заболевания — двусторонние диссеминированные изменения ретикулярного или ретикулонодулярного характера, более выраженные в нижних отделах легких.
На ранних этапах развития заболевания может наблюдаться уменьшение объема легочных полей и понижение прозрачности легких по типу «матового стекла», данные изменения особенно заметны при сравнении нескольких рентгенограмм пациента. При прогрессировании заболевания ретикулярный паттерн становится более грубым, появляются округлые кистозные просветления размером 0,5–2 см, отражающие формирование «сотового легкого», могут быть видны линейные тени дисковидных ателектазов. Рентгенологическая картина при ИФА может коррелировать с гистопатологическими изменениями, однако такая связь существует лишь при наличии признаков «сотового легкого». На поздних стадиях ИФА рентгенологическая картина часто выявляет девиацию трахеи вправо, трахеомегалию. Следует обратить внимание, что около 15 % пациентов с гистологически доказанным диагнозом ИФА могут иметь неизмененную рентгенологическую картину; это позволит избежать большого количества диагностических ошибок. Вовлечение плевры, внутригрудная аденопатия, локализованные паренхиматозные уплотнения не характерны для ИФА и могут отражать либо другое интерстициальное заболевание легких, либо осложнения заболевания, такие как инфекции или опухоли.
Более ценную информацию можно получить при помощи компьютерной томографии (КТ), особенно КТ высокого разрешения (ВРКТ). Компьютерная томография значительно обогатила наши представления о частоте, характере полостных образований в легких и их роли в диагностике различных заболеваний. С появлением КТ, особенно спиральной высокоразрешающей, полостные образования в легких стали выявляться гораздо чаще. Причем проблема полостных образований легких приобрела особое значение в плане их дифференциальной диагностики, патогенеза и понимания сущности тех заболеваний, на фоне которых они развиваются.
Характерными находками при КТ являются нерегулярные линейные тени, снижение прозрачности легочных полей по типу «матового стекла» и кистозные просветления размером от 2 до 20 мм в диаметре. Признаки «сотового легкого» при КТ выявляются значительно чаще по сравнению с обзорной рентгенографией легких.
Наибольшие изменения выявляют в базальных и субплевральных отделах. КТ-паттерн и распределение изменений в большинстве случаев являются патогномоничными для ИФА. Кроме того, КТ отражает морфологические признаки ИФА: ретикулярный паттерн соответствует фиброзу, а паттерн «матового стекла» — клеточной инфильтрации. Картина «матового стекла» проявляется только при минимальном утолщении альвеолярных стенок, интерстиция или частичном заполнении альвеол клетками, жидкостью, аморфным материалом. Изменения являются двусторонними, зоны «матового стекла» в большинстве случаев имеют распространенный характер. Утолщение междольковых перегородок, равномерное утолщение стенок бронхов, расширение тени сосудов наблюдаются преимущественно в средних и нижних зонах легких. Субплевральная локализация характерна для утолщения междольковых перегородок и «матового стекла». Равномерное утолщение стенок бронхов и расширение сосудистых теней прослеживаются во всех случаях центрально в задних сегментах средненижних зон.
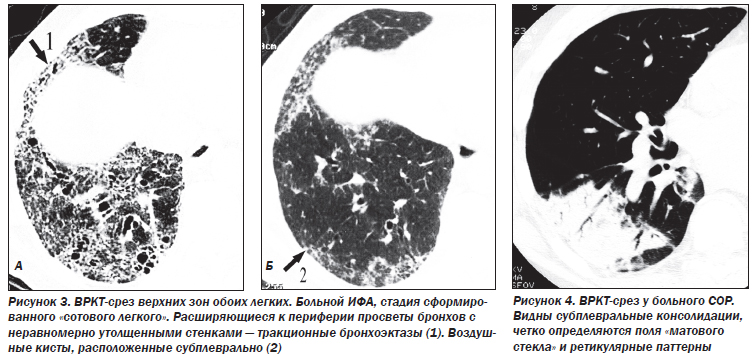
К поздним стадиям ИФА относятся случаи, когда патологический процесс представлен фиброзными изменениями различной степени выраженности. В зависимости от характера фиброзных изменений патологический процесс подразделяется на этапы интерстициального фиброза, формирования «сотового легкого» и сформированного «сотового легкого» с признаками альвеолита или без них. В основу положены критерии: деформация структур интерстициальной легочной ткани, уменьшение объема легочной ткани (доли, долей или целого легкого).
Деформация легочного рисунка, являющаяся маркером фиброзных изменений в легочной ткани при ИФА, представлена следующими симптомами: участки уплотнения легочной ткани, линейные и/или сетчатые тени, не являющиеся междольковыми перегородками; воздушные кисты диаметром 2–20 мм с толстыми (> 2 мм) стенками; расширение терминальных бронхиол — бронхиолоэктазы; неравномерное расширение субсегментарных и сегментарных бронхов — тракционные бронхоэктазы; сочетание расширения бронхососудистых пучков и неравномерного утолщения стенки бронхов и сосудов.
Фокусы уплотнения легочной ткани являются признаком частично обратимого процесса. Сетчатая и/или линейная деформация легочного рисунка — признак необратимого интерстициального фиброза линейной формы. Воздушные кисты диаметром 2–20 мм, бронхиолоэктазы, тракционные бронхоэктазы, сочетание расширения бронхососудистых пучков и неравномерного утолщения стенки бронхов и сосудов — элементы сформированного необратимого «сотового легкого».
Наряду с признаками, характеризующими необратимые фиброзные изменения в легочной ткани, как правило, определяются элементы активного обратимого патологического процесса в виде утолщения междольковых перегородок и участков понижения прозрачности легочной ткани по типу «матового стекла».
Перфузионная сцинтиграфия с радиоактивным технецием легких позволяет оценить степень поражения легочного кровотока на уровне мелких сосудов и капилляров у больных с ИФА.
КТ-картина имеет прогностическое значение. Считается, что лучший прогноз имеют пациенты с паттерном «матового стекла», худший — с ретикулярным паттерном и промежуточный — со смешанным паттерном. В настоящее время по предсказательной ценности КТ выходит на первый план, опережая функциональные легочные тесты, бронхоальвеолярный лаваж и даже биoпсию легких, так как позволяет дать оценку поражения практически всей паренхимы легких по сравнению с отдельным биопсийным образцом. При сочетании ИФА с эмфиземой КТ является единственным методом, позволяющим оценить выраженность эмфиземы, которая преимущественно локализуется в верхних отделах, и разграничить ее с кистозными изменениями, характеризующими «сотовое легкое».