Резюме
Атиповий гемолітико-уремічний синдром (aГУС) є надзвичайно рідкісним, але небезпечним для життя захворюванням у дітей через те, що може спричинити гостре пошкодження нирок. Пацієнти з aГУС мають ризик розвитку повторних епізодів. Отже, у цій статті ми представляємо випадок рецидиву аГУС у хлопчика 9 років. Дитина надійшла у відділення невідкладної допомоги на п’ятий день хвороби з основними скаргами на одутлість обличчя та зменшення виділення сечі. З анамнезу відомо, що в пацієнта розвинувся другий епізод aГУС через шість років після повного одужання від першого епізоду. Клінічні прояви обох епізодів аГУС були спровоковані інфекцією дихальних шляхів, гастроінтестинальні симптоми не відмічалися. Результати діагностичних досліджень, проведених під час першого епізоду аГУС, наступні: аналіз калу на Escherichia coli й токсини Shiga був негативним; дослідження комплементу не виявили порушень; активність ADAMTS13 і рівень антитіл до фактора Н комплементу були в нормі. Результати ультразвукового дослідження та біопсії нирок відповідали діагнозу. Сімейний анамнез характеризувався діагнозом аГУС у молодшого брата пацієнта, що було підтверджено молекулярно-генетичним тестуванням, зокрема виявлено патогенний варіант у гені CD46/MCP (мембранний кофакторний білок) у гетерозиготному стані. При фізикальному обстеженні зафіксовано блідість, набряк обличчя, помірну артеріальну гіпертензію, олігурію. Лабораторно виявлено гемолітичну анемію, тромбоцитопенію, значну азотемію, різке зниження клубочкової фільтрації, високий рівень аспартатамінотрансферази, незначний електролітний дисбаланс, протеїнурію. Підтримуюче лікування включало інфузійну терапію, використання свіжозамороженої плазми, фуросеміду й дексаметазону. Дитині розпочато гострий гемодіаліз унаслідок тяжкого гострого пошкодження нирок. Висновки. Рецидив аГУС характеризується тяжкою нирковою недостатністю, що потребує гострого гемодіалізу. Вірусні інфекції є потенційними тригерами аГУС. Повторний перебіг захворювання та позитивний сімейний анамнез вказують на важливість генетичного скринінгу, оскільки слід розглядати наявність сімейного аГУС.
Background. Atypical hemolytic uremic syndrome (aHUS) is an extremely rare but life-threatening disorder in children since it may cause acute kidney injury. Patients with aHUS are at risk of recurrence. Hence, in this paper, we present a case of a 9-year-old boy with aHUS relapse. The child was admitted to the emergency department on the fifth day of illness with main complaints of facial puffiness and decreased urine output. Based on the medical history, the patient developed the second episode of aHUS after 6 years of complete recovery from the first episode. There was no preceding diarrheal illness, instead, the clinical manifestation of both aHUS episodes was triggered by a respiratory tract infection. The results of diagnostic studies performed during the first episode of aHUS were as follows: stool tests for Escherichia coli and Shiga toxins were negative; a complement assay showed no abnormalities; ADAMTS13 activity and anti-complement factor H antibodies were normal. The results of the kidney ultrasonography and biopsy were consistent with the diagnosis. Family history was remarkable for aHUS in a younger sibling confirmed by molecular genetic testing, in particular, a pathogenic variant in the CD46/MCP (membrane cofactor protein) gene in the heterozygous state has been identified. Physical examination revealed paleness, facial swelling, moderate hypertension, and oliguria. Laboratory findings demonstrated hemolytic anemia, thrombocytopenia, significant azotemia, a severe reduction in the glomerular filtration rate, a high level of aspartate aminotransferase, insignificant electrolyte imbalance, and proteinuria. Supportive treatment included fluid and electrolyte management, fresh frozen plasma, furosemide, and dexamethasone. The child commenced acute hemodialysis due to severe acute kidney injury. Conclusions. A recurrence of aHUS is characterized by severe renal failure requiring acute hemodialysis. Viral infections are potential triggers of aHUS. A relapsing course of the disease and a family history of aHUS indicate the importance of genetic screening, as familial aHUS should be considered.
Introduction
Atypical hemolytic uremic syndrome (aHUS) is a rare thrombotic microangiopathy characterized by hemolytic anemia, thrombocytopenia, and acute kidney injury. The current prevalence of aHUS among children remains unclear. Based on available studies, the annual incidence rate ranges between 0.26 and 0.75 per one million [1], and the age of onset varies from the neonatal period to adulthood [2]. Cases of aHUS are not associated with Shiga toxin-producing Escherichia coli, but rather caused by dysregulation of the alternative complement pathway due to complement gene mutations or anti-complement factor H (CFH) autoantibodies [3, 4]. Pediatric aHUS patients frequently experience relapse. Abnormal kidney function tests are one of the major laboratory findings. Renal involvement ranges from hematuria and proteinuria to severe renal failure [5]. Clinical manifestation of aHUS episodes is most commonly preceded by viral infections [6]. Progression of renal impairment to end-stage renal disease occurs in 50–60 % of patients [2, 7]. The diagnosis of aHUS is basically made after excluding Shiga toxin-producing Escherichia coli, thrombotic thrombocytopenic purpura, and secondary hemolytic uremic syndrome associated with systemic diseases [8]. Besides clinical and laboratory features, examination of patients with aHUS includes biochemical assessment of the complement alternative pathway, determination of anti-CFH antibodies, and the last step is genetic screening [5, 9]. However, the capacity for genetic screening in most developing countries is limited [8].
Initial management of aHUS patients is usually based on clinical presentation and available laboratory studies because complement assay and genetic analysis take a longer time to be completed [5, 8]. Plasma therapy still remains the mainstay of management for aHUS since up-to-date treatment options (e.g., eculizumab, ravulizumab) may not be available in resource-limited countries [2–4].
Atypical HUS can be sporadic or familial. Familial aHUS accounts for about 20 % of all cases and should be recognized in the presence of aHUS in at least two members of the same family with diagnoses at least 6 months apart. Genetic abnormalities in complement genes are found in approximately 70 % of patients [10]. Hence, in this paper, we present a case of a 9-year-old boy diagnosed with a se–cond episode of aHUS triggered by a viral infection and associated with a family history of aHUS. This case is significant due to its rarity and therefore may help improve understanding of the course of the disease and analyze avai–lable diagnostic and therapeutic options.
Case report
A 9-year-old male presented to the emergency department with facial puffiness and reduced urine output. Five days prior to admission, he developed a fever of 38.3 °C, dry cough and sore throat. Three days prior to admission, dark urine appeared; the boy was seen by a nephrologist in the outpatient department, rehydration therapy and ibuprofen were recommended. Despite this management, his condition worsened due to a gradual decrease in urine output and therefore he was admitted to our City Children’s Hospital.
Based on his medical history, the child first experienced aHUS accompanied by mild renal involvement (maximum serum creatinine of 155 µmol/L) at the age of 3 years. Initial symptoms were also preceded by an upper respiratory tract infection; no diarrheal illness was noted at that time. Besides the classic triad of symptoms, the diagnosis of aHUS was based on the following results: stool assay for enterohemorrhagic Escherichia coli PCR and Shiga 1 and 2 toxins showed negative results; significant non-selective glomerular proteinuria of 2212 mg/m2/day; kidney ultrasound revealed bilateral increased parenchymal echogenicity with dimi–nished corticomedullary differentiation; renal biopsy showed thrombotic microangiopathy and acute tubular necrosis. Complement analysis included functional assay (total hemolytic complement (CH50) and complement alternative pathway (AP50)), quantification of complement components and regulators (complement C3, C4 plasma levels, CFH and complement factor I (CFI)). However, complement anomalies were not detected. Anti-CFH antibodies and ADAMTS13 (a disintegrin and metalloproteinase with thrombospondin type 1 motif 13) activity were normal. Supportive treatment resulted in a significant improvement in kidney function tests, urine protein excretion decreased to 215 mg/m2/day. A low dose of ramipril was recommended for subsequent treatment. The patient has not had recurrent symptoms of renal impairment and hemolysis for 6 years. Apart from that, he had no underlying diseases.
Family history showed that the brother of our patient, who is one year younger, developed an episode of aHUS at the age of 5 years. Molecular genetic analysis of a younger sibling demonstrated that he carried the pathogenic splice site variant c.286+2T>G p. (Lys49_Arg96del) in the CD46/MCP (membrane cofactor protein) gene in the hete–rozygous state.
On admission, the boy appeared pale and edematous. His level of consciousness was 15 points on Glasgow Coma Scale. Pupils were equally round and reactive to light. Me–ningeal signs were negative. There was no jaundice or petechial rash. His body temperature was normal, weight was between the 50th and 85th percentile. Vital signs: body temperature 36.8 °C, respiratory rate 18/min, heart rate 78/min, blood pressure 130/80 mmHg, oxygen saturation 96–98 % on room air. Capillary refill time was less than 2 seconds. Cardiac, pulmonary, and abdominal findings were unremarkable. Urine output was 100–120 ml/24 h.
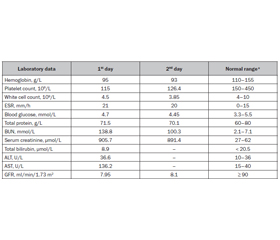
Baseline workup revealed a hemoglobin level of 95 g/L, a hematocrit value of 29 %, a platelet count of 115 × 109/L, a creatinine level of 905.7 µmol/L, an elevated blood urea nitrogen (BUN) of 138.8 mmol/L, and a high aspartate aminotransferase (AST) level of 136.2 U/L. Peripheral blood smear showed schistocytes. Blood pH, serum potassium, sodium, and ionized calcium levels were 7.3; 4.28; 128.3, and 0.67 mmol/L, respectively. Prothrombin and partial thromboplastin time were 14.3 and 22.2 seconds, respectively, and the international normalized ratio was 1.40. A urinalysis revealed red blood cells of 5–6/hpf, single granular casts, and protein of 1.85 g/L. C-reactive protein was 6 mg/L. Indirect Coombs test was negative. The results of laboratory studies were summarized in Table 1. The electrocardiogram revealed a prolonged QT interval. Echocardiography was unremarkable, whereas renal ultrasonography showed slightly enlarged, hyperechogenic kidneys. No enterohemorrhagic Escherichia coli was detected by culture later on.
/93.jpg)
Considering the aforementioned, the patient was diagnosed with aHUS (second episode) and managed symptomatically with adequate hydration (intravenous sterofundin ISO, oral rehydration solution), infusion of fresh frozen plasma (FFP), furosemide, dexamethasone, and lactulose. On the second day of admission, we transferred the patient to another hospital capable of initiating acute hemodialysis (HD) due to marked azotemia, oliguria (urine output was 0.2 ml/kg/h), and GFR below 10 ml/min/1.73 m2. The patient’s condition gradually improved with the normalization of renal function tests, urinalysis and complete blood count. We recommended genetic testing of the patient.
Discussion
The present case demonstrates the child with a severe relapse of aHUS. The hallmark of the second aHUS episode was severe acute kidney injury requiring acute HD. In addition to renal impairment, hemolytic anemia, thrombocytopenia, and a negative Shiga toxin-producing Escherichia coli culture represented the diagnostic criteria for aHUS. Also, moderate hypertension was present. In recent years, the term aHUS has been used to designate complement-mediated HUS caused by dysregulation of the alternative complement pathway due to pathogenic genetic variants or antibodies to components or regulatory factors in this pathway [4, 11]. However, causes of aHUS may also include inborn errors of cobalamin C metabolism and diacylglycerol kinase epsilon gene mutations [12]. HUS linked to autoimmune diseases, transplantation, malignancies, or certain drugs is often defined as secondary, mainly non-relapsing form of HUS [8, 13]. Thus, it has been proposed to apply the term aHUS to patients without coexisting diseases [14]. In the pediatric group, aHUS accounts for 5–10 % of all HUS cases [3]. Generally, aHUS should be distinguished from thrombotic thrombocytopenic purpura by evaluating ADAMTS13 activity. Patients with thrombotic thrombocytopenic purpura have abnormally low ADAMTS13 activity (usually must be < 10 % of normal) in contrast to aHUS cases [5].
In our patient, both episodes of aHUS followed a respiratory illness. We did not routinely detect a pathogen since the child’s respiratory symptoms had subsided by the time of admission. Several viral infections may induce clinical ma–nifestation and relapse of aHUS, in particular Epstein-Barr virus, varicella, human immunodeficiency virus, influenza A virus, parvovirus B19, and Coxsackie virus [6, 8]. Moreover, recent studies have shown that the clinical symptoms of aHUS in children were triggered by influenza B virus and SARS-CoV-2 infection. Hence, influenza and COVID-19 vaccination may be beneficial to protect patients at risk [6, 7, 15, 16].
Primary investigation of the complement status of the patient was essential and aimed to identify complement-mediated HUS. Sridharan et al. recognized a decrease in both complement factor B (CFB) and CH50 as an important indicator to confirm the diagnosis [17]. However, in the pre–sent case, basic serological complement biomarkers (CH50, AP50, C3, C4, CFH, CFI) as well as anti-CFH antibodies were unremarkable. Loirat et al. found that decreased C3 levels were observed in only 30–40 % of patients with aHUS and normal complement plasma levels (e.g., CFH or CFI) don’t exclude a mutation in the corresponding gene. All patients with suspected aHUS should be tested for the pre–sence of anti-CFH antibodies [14], although positive results have been reported in no more than 20 % of patients [18].
Nowadays, genetic screening plays a dominant role in diagnosing patients with aHUS in developed countries. Genetic variants in complement regulatory proteins (CFH, CD46/MPC, CFI, C3, CFB, thrombomodulin gene (THBD)) are present in 50–60 % of aHUS patients [7]. CFH and CD46/MPC variants appear to be the most common genetic abnormalities in patients with complement-mediated HUS and present in approximately 20–30 and 5–15 % of cases, respectively [19]. Notably, nine out of ten patients demonstrate a heterozygous state of genetic variants [12]. Prior research has reported an association between genetic abnormalities affecting the complement system and the clinical course, recurrence and outcome of aHUS. Patients with CD46/MCP mutations frequently experience relapses followed by a complete recovery. This category of patients generally has a normal serum C3 level and better long-run outcome, with about 30 % progressing to end-stage renal disease [14]. In contrast, children with the CFH pathogenic variant have a high incidence of recurrence with approximately a 60–80% rate of end-stage renal disease [2].
This case report highlights the importance of genetic testing, taking into account the current relapse of aHUS, a previous episode followed by a full recovery, and a positive family history of aHUS (a sibling has been diagnosed with aHUS associated with a heterozygous pathogenic variant in the CD46/MCP gene). In particular, the patient should undergo genetic analysis of the minimum set of genes, including CFH, CFI, CD46/MCP, CFB, THBD, complement factor H-related 1 and 5, and C3 [11]. Familial aHUS should be highly suspected in this case. Previous studies reported that siblings of aHUS patients are at a greater di–sease risk than parents, although any family member may be a healthy gene carrier [20]. The principal concern remains that genetic screening in developing countries might not be available immediately or be inaccessible at all.
Supportive therapy was provided to the patient as the mainstay of initial treatment for aHUS. Appropriate fluid and electrolyte management was based on careful clinical and laboratory assessment as mild hyponatremia and hypocalcemia were observed. Infusion of FFP is available worldwide and remains an important treatment for aHUS, even though some authors have indicated a lack of evidence for plasma therapy effectiveness in aHUS [2, 21]. Preferably, plasma therapy should be switched to eculizumab (a human anti-C5 monoclonal antibody) for subsequent therapy [14]. Eculizumab showed excellent efficacy in treating patients with CFH and CFI genetic variants and appeared to be a beneficial treatment for anti-CFH antibody-associated HUS [22]. According to the current recommendations, genetic screening results are not necessary to initiate treatment with eculizumab since patients with and without identified gene mutations in complement factors demonstrate similar response to this therapy [4]. However, eculizumab may not be widely accessible in resource-limited countries and therefore plasma therapy is often the best treatment option, in particular in the present case. And last but not least, in this case, acute HD was applied due to the continued deterioration of renal function. Fortunately, kidney function tests returned to normal values after discontinuation of HD.
Conclusions
Pediatric aHUS is an extremely rare disorder that may have a relapsing course and lead to life-threatening acute kidney injury requiring renal replacement therapy. A child can remain asymptomatic between episodes even for some years. Most commonly, respiratory infections trigger the onset of aHUS episodes. Indeed, continuous follow-up is an important component of improving patient outcomes. Furthermore, timely access to medical care is critical, as any delay in seeking medical attention increases the likelihood of an unfavorable clinical course. Infusion of FFP is an effective available treatment for aHUS.
Comprehensive complement investigation is essential to identify potential complement anomalies, although a normal complement profile does not rule out the diagnosis of aHUS. Molecular genetic analysis to detect pathogenic variants in the genes associated with aHUS can be beneficial to a patient with repeated episodes and positive family history, not only to confirm the diagnosis but also for subsequent approaches to management. Familial aHUS may reasonably be presumed in this case, as two members of the same family are diagnosed with aHUS.
Received 02.04.2023
Revised 12.04.2023
Accepted 18.04.2023

/93.jpg)