
Газета «Новости медицины и фармации» Нефрология (297) 2009 (тематический номер)
Вернуться к номеру
Захворювання органів сечової системи у дітей та синдром дисплазії сполучної тканини
Авторы: І.В. Багдасарова, Т.В. Буднік, А.В. Малахова, Інститут нефрології АМН України, відділ дитячої нефрології, м. Київ
Луганський державний медичний університет, кафедра педіатрії ФПО, Луганська обласна дитяча клінічна лікарня
Версия для печати
Вступ
На сучасному етапі актуальними в дитячій нефрології постають питання не лише прогресуючої поширеності захворювань органів сечової системи (ОСС), а й частішання факту нетипового їх перебігу, як то прогредієнтність, стероїдорезистентність, тривала малосимптомність з маніфестацією хронічної ниркової недостатності (ХНН).
У зв''язку з цим науковцями переглядаються подання про набуті захворювання мультифакторної природи, з''являються нові концепції щодо сутності хвороби, доповнення факторів ризику й схильності до хвороби, розглядаються гіпотези про сукупність несприятливих екзогенних та ендогенних факторів, що реалізують «програму» хвороби на всіх рівнях організму починаючи з клітинного [1, 3].
У світлі викладеного залишається незакритим питання про роль у цьому спадково обумовленої неспроможності колагенової структури, тобто дисплазії сполучної тканини (ДСТ), ознаки якої все з більшою сталістю зустрічаються також і серед хворих нефрологічної групи та істотно впливають на характер і перебіг захворювання [2, 3].
За сучасним поданням синдром ДСТ (СДСТ) був відокремлений як самостійна нозологічна форма в 1990 р. (м. Омськ, Росія). ДСТ розподіляють на диференційовані (ДДСТ) та недиференційовані (НДСТ). Диференційовані ДСТ характеризуються певним типом успадкування та чіткою клінічною картиною. Недиференційовані ДСТ діагностують, коли в пацієнта набір клінічних ознак не вкладається в жодне спадкове моногенне захворювання.
За Т.І. Кадуриною, НДСТ — генетично гетерогенна група захворювань мультифакторної природи з прогредієнтним перебігом, в основі якого лежать порушення синтезу, розпаду чи морфогенезу компонентів позаклітинного матриксу, що виникає в період раннього ембріогенезу чи постнатально під впливом несприятливих факторів оточуючого середовища й може виявитися в різні періоди життя [4].
Очевидно, що НДСТ можуть бути причиною диспластичних змін сполучної тканини різних органів і систем та відповідно основою формування різних хронічних захворювань.
Етіопатогенез уродженої неповноцінності сполучної тканини у світі до кінця не вивчений, але дані численних досліджень показують, що клінічні прояви ДСТ обумовлені аномалією колагенових структур. На сьогодні відкрито 19 генетичних типів колагенів. Кожна тканина характеризується строгим набором, будовою та співвідношенням типів колагену [5].
Уроджені дефекти фібрилогенезу, за який відповідає колаген IV типу, сприяють формуванню різних варіантів патології нирок та сечових шляхів. Серед ДДСТ з ураженням органів сечовиділення: синдром Альпорта, сімейна доброякісна гематурія, дифузний лейоміоматоз стравоходу та геніталій, синдром Фразера, синдром мікрокорнеа — уроджений нефроз Пірсона, тромбоцитопенічна пурпура, гіпофосфатемічний рахіт та ін. [4].
На цей час у літературі існує порівняно небагато повідомлень стосовно сполучення симптомів ДСТ та проявів захворювання з боку органів сечовиділення. Зроблено акцент на зв''язок нефроптозу та дистопії нирок, дисплазії сечоводів та статевих органів, рефлюкс-нефропатії, дисплазії та гіпоплазії ниркової паренхіми з фенотипічними проявами недиференційованої дисплазії сполучної тканини [1, 2]. За деякими даними, вважається, що ураження верхніх сечових шляхів при генетичних дефектах сполучної тканини порівнянне з поширеністю малих аномалій розвитку серця (МАРС), що прийнято вважати маркером недиференційованої дисплазії сполучної тканини [2, 6]. Ряд авторів указують на резистентність до стандартної терапії, рецидивуючий перебіг, раннє зниження функцій нирок серед хворих на пієлонефрит, гломерулонефрит на фоні недостатності сполучної тканини [1–3].
Тому метою нашого дослідження стало вивчення частоти виявлення синдрому ДСТ та характеру перебігу захворювання залежно від його наявності серед дітей із захворюваннями ОСС.
Об''єкт та методи дослідження
У дослідження було включено 308 дітей із різними захворюваннями ОСС, асоційованими з ДСТ та без неї, які знаходились на стаціонарному лікуванні на нефрологічних ліжках Луганської обласної дитячої клінічної лікарні у 2008 р. Верифікація діагнозу відбувалася згідно зі стандартними загальноприйнятими методами клінічного, лабораторного та інструментального обстеження з консультацією генетика, а також були застосовані експертні таблиці з бальною оцінкою значимості цілого ряду клінічних, лабораторних та інструментальних показників ДСТ, рекомендовані Т.І. Кадуриною (2009) [4]. Згідно з першою експертною таблицею значимості фенотипічних та клінічних маркерів в оцінці ступеня вираженості, ДСТ I (легкому) ступеню відповідала кількість балів < 12, ІІ (середньому) — < 23 балів, ІІІ (тяжкому) — > 24 балів. За другою експертною таблицею значимості клініко-інструментальних і лабораторних показників у діагностиці тяжкості ДСТ І ступеня відповідала сума балів до 20, ІІ ступеня — 21–40 балів, ІІІ ступеня — 41 бал і більше.
При обробці результатів дослідження використовувалися стандартні засоби Microsoft Excel 2007. Для оцінки шансів розвитку патологічних станів використовували критерій χ2 для чотирипільної таблиці сполученості з корекцією на безперервність за Йєйтсом з розрахунком стандартної похибки оцінки шансів (SE(о)) та її довірчого інтервалу.
Результати та їх обговорення
Серед обстежених СДСТ був наявним у 72 % (216/308) випадків (р < 0,05). Частота СДСТ при різних захворюваннях ОСС варіювала. Диференційовані ДСТ з ураженням органів сечовиділення (синдром Альпорта, сімейна доброякісна гематурія, гіпофосфатемічний рахіт) становили 13 % (28/216).
У хворих із малими аномаліями нирок (МАН) частота СДСТ становила 69 % (80/116), з нейрогенною дисфункцією сечового міхура (НДСМ) — 78 % (45/58), з гострим пієлонефритом (ГПН) — 72 % (102/142), з гострим гломерулонефритом (ГГН) — 87 % (61/70) та найчастіше серед хворих на хронічний пієлонефрит (ХПН) — 100 % (68/68) та хронічний гломерулонефрит (ХГН) — 100 % (52/52) (рис. 1).
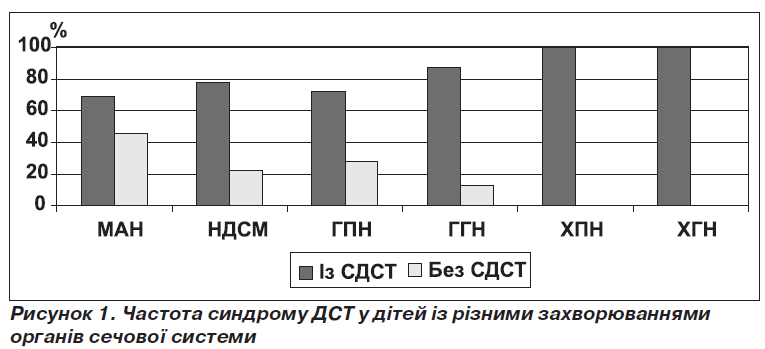
За даними статистичного аналізу, відношення шансів виникнення МАН у дітей із проявами ДСТ та без них суттєво не відрізняється й дорівнює 0,92 (SE(о) = 0,23, довірчий інтервал 0,55–1,5; рχ2 < 0,05); відношення шансів виникнення НДСМ відрізняється незначно — 1,6 (SE(о) = 0,55, довірчий інтервал 0,8–3,03; рχ2 < 0,05); відношення шансів виникнення ГПН дорівнює 1,16 (SE(о) = 0,29, довірчий інтервал 0,71–1,89; рχ2 < 0,05); відношення шансів виникнення ГГН помітно відрізняється — 3,69 (SE(о) = 1,39, довірчий інтервал 1,66–7,2; рχ2 < 0,05).
Кількість осіб жіночої статі з проявами СДСТ при захворюваннях ОСС була відчутно більшою — 64,8 % (140/216).
Патології з боку сосудів та серця супроводжували перебіг захворювань ОСС у вигляді малих аномалій розвитку серця у 62 % (134/216), захворювання ШКТ у вигляді дискінезії жовчовивідних шляхів (ДЖВШ) — у 53 % (114/216), різні види порушення статури та плоскостопість (ПКС) — у 38 % (82/216), з боку нервової системи (ПНС) частіше зустрічалися порушення у вигляді ВСД — 72 % (155/216), з боку імунної системи (ПІС) – рецидивуючі вірусні та/або бактеріальні інфекції — у 82 % (177/216), патологія розвитку зовнішніх статевих органів (ПЗСО) — у 15 % (32/216) та інші прояви, що ще раз підкреслили причетність факту порушення обміну сполучної тканини серед означеного контингенту дітей (рис. 2).
Використання експертних таблиць дозволило нам не лише виявити значну кількість ознак або мікроознак дизморфогенезу та цілий каскад захворювань з боку інших органів та систем, а й отриманий індекс «синдроміальності» співвіднести зі ступенем тяжкості перебігу основного захворювання (рис. 3).
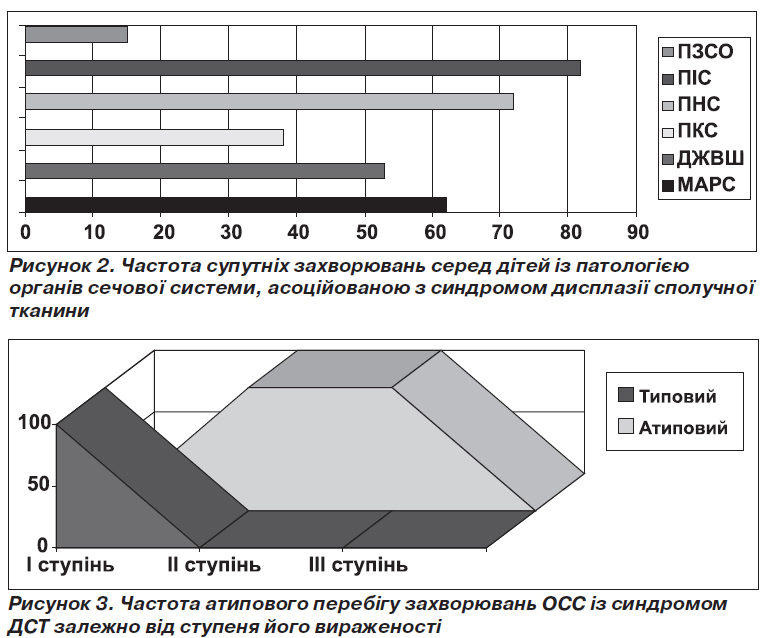
Прогредієнтний перебіг захворювання, резистентність до стандартної терапії мали місце у 27 % (58/216) хворих із проявами СДСТ. Причому в 100 % (58/58) випадків це були хворі з ІІ–ІІІ ступенем вираженості СДСТ.
Висновки
Отримані дані переконливо свідчать про поширеність СДСТ серед дітей із захворюваннями ОСС, його безсумнівний вплив на перебіг захворювання та прогноз, змушують з іншого боку поглянути на патогенетичні й терапевтичні аспекти ведення таких хворих; приділяти увагу ранній верифікації цього стану серед нефрологічного контингенту дітей.
1. Игнатова М.С., Шатохина О.В. Клинико-генетические аспекты диагностики нефропатий у детей // Нефрология и диализ. — 2003. — 5. — 1.
2. Инзель Т.Н., Гаглоева Т.М., Ковальський С.В. Диагностическое значение специфических фенотипических маркеров аномалий развития почек, ассоциированных с синдромом дисплазии соединительной ткани // Урология. — 2000. — 3. — 8-12.
3. Цыгин А.Н. Патогенетические основы первичного нефротического синдрома и лечение его стероидорезистентных вариантов у детей: Дис… д-ра мед. наук. — М., 1996. — 198 с.
4. Кадурина Т.И., Горбунова В.Н. Дисплазия соединительной ткани: Руководство для врачей. — СПб.: Элби-СПб., 2009. — 704.
5. Beighton P., Paepe A., Steinmann B. International nosology of heritable disorders of connective tissue // Am. J. Med. Genet. — 1998. — Vol. 77, № 2. — P. 31-37.
6. Boudoulas H., Kolibach A., Baker P. et al. Mitral valve prolaps and the mitral valve prolaps syndrome: a diagnostic classification and pathogenesis of symptoms // Amer. Heart J. — 1989. — Vol. 118, № 4. — P. 796-818.