
Газета «Новости медицины и фармации» Гастроэнтерология (304) 2009 (тематический номер)
Вернуться к номеру
Наследственные нарушения углеводного обмена, ведущие к поражению печени
Авторы: К.Н. Бородий, Донецкий национальный медицинский университет им. М. Горького
Версия для печати
К патологии печени приводит гетерогенная группа наследственных болезней, обусловленных различными видами нарушения обмена углеводов. Различают:
Нарушения обмена моно- и дисахаридов
Фруктоземия
Этиология и патогенез. Заболевание обусловлено врожденным отсутствием ферментов фруктозофосфатальдолазы и фруктозодифосфатальдолазы. Избыточное накопление фруктозофосфата нарушает гликогенолиз, что приводит к гипогликемии. В печени имеется недостаточное количество фермента фруктозо-1-фосфат-альдолазы, в результате продукты обмена (фруктозо-1-фосфат) накапливаются в организме (печени, почках, слизистых оболочках кишечника) и оказывают повреждающее действие. Морфологически в печени выявляются жировая инфильтрация, умеренный перилобулярный фиброз.
Клиническая картина. Симптомы возникают при введении в рацион сладкой пищи или фруктовых соков, т.е. продуктов, содержащих фруктозу. Со 2–4-го месяца развиваются диспептические явления и состояния острой гипогликемии, которые проявляются бледностью, вялостью, потливостью, запахом ацетона. В тяжелых случаях может развиться гипогликемическая кома с потерей сознания и судорогами. Характерно, что гипогликемия возникает после приема пищи. С возрастом дети сами отказываются от сладкой пищи. Постоянным признаком является гепатомегалия (обычно с увеличением обеих долей) с ровным краем и некоторым уплотнением печени.
Лечение. Диета, лишенная фруктозы, главными источниками которой считаются мед, сахарный тростник и свекла, фрукты, джемы, повидло, морковь, какао, цикорий, репа. Больным разрешается употреблять молоко и молочные продукты, яйца, маслины, подсолнечное масло, животные жиры. Разрешается женское и коровье молоко. Сухое молоко должно быть без сахара (сахарозы). Допускаются все виды сыров и натуральные кисломолочные продукты (неподслащенные). Запрещается молоко с добавлением сахарозы, сгущенное молоко, подслащенные кисломолочные продукты. Разрешены мясо и рыба. Исключаются колбасы и колбасные изделия, консервы. Жиры включаются в диету практически без ограничений (сливочное и растительное масло, маргарин). Почти все фрукты запрещены. Можно употреблять в пищу лимоны и каштаны. Из овощей допускаются зеленые бобы, кресс-салат, латук, лук-порей, капуста, шпинат. Разрешаются натуральная пшеничная или ржаная мука, рис, хлеб, манная крупа, чай, кофе, какао без сахара, глюкоза, мальтоза, декстрин-мальтоза, сахарин. Запрещаются соя, мука с сахарозой, бисквиты, пирожные, лимонад и все газированные фруктовые напитки, соки, сиропы, сахар, варенье, нуга, конфеты, все медикаменты, содержащие сахар, сорбитол (гранулы, драже, порошки, пилюли).
Новорожденным назначают молочные смеси без сахара. Дети первого года получают молочные смеси, содержащие только лактозу и декстрин-мальтозу. Вместо фруктовых пюре и соков питание дополняют глюкозой (от 30 до
Галактоземия
Частота 1 : 35 000–50 000 населения.
Этиология и патогенез. Наследственная энзимопатия. Наследуется по рецессивному типу. В основе галактоземии (рис. 1) лежит нарушение обмена галактозы в связи с отсутствием фермента галактозофосфат-уридилтрансферазы. В результате в крови накапливаются в больших концентрациях галактоза и галактозофосфат. Происходит нарушение процесса ферментативного превращения галактозы в глюкозу с накоплением галактозы и продуктов ее обмена в клетках, что оказывает повреждающее действие на функции печени, головного мозга, хрусталика глаза, почек.
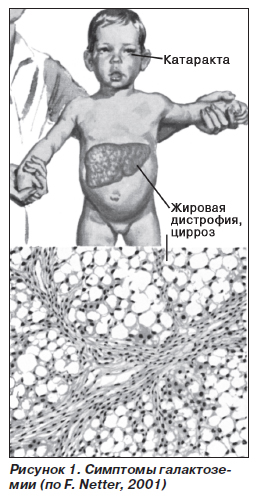
Диагностика:
1. Определение концентрации га-лактозо-1-фосфата в эритроцитах (повышена).
2. Исследование активности галак-тозо-1-фосфат-уридилтрансферазы в эритроцитах.
3. Повышение уровня галактозы в крови и моче (методом хроматографии).
4. Микробиологический тест Гатри.
Лечение. Диетотерапия является единственным методом лечения. Для вскармливания ребенка используют смеси, лишенные лактозы. Из питания детей более старшего возраста исключают цельномолочные продукты.
Гликогенозы
Этиология и патогенез. Группа наследственных болезней обмена полисахаридов, развивающихся в результате нарушения синтеза или распада гликогена на простые сахара. При этом нормальный и аномальный гликоген одновременно накапливаются в клетках печени и других органах и тканях.
Гликоген — это сильно разветвленный полимер глюкозы, в котором большинство остатков имеют 1,4-связи, а 7–10 % остатков — 1,6-связи. Древовидная структура подвергается надстройке и отщеплению остатков на периферии молекулы. Молекулярная масса гликогена составляет несколько миллионов, его молекулы могут агрегировать с образованием структур, видимых при электронной микроскопии. В печени гликогена обычно содержится менее 70 мг/г, а в мышцах — менее 15 мг/г, но эти величины колеблются в зависимости от питания и гормональных влияний. Нарушения структуры гликогена могут быть связаны как с уменьшением, так и с увеличением ветвления молекулы.
Метаболические пути синтеза и распада гликогена в разных тканях различны; например, некоторые реакции активно протекают в печени, но менее активны или отсутствуют в мышцах, а ряд ферментов в мышцах и печени кодируются разными генами. Глюкоза плазмы проникает в клетку и фосфорилируется глюкокиназой или гексокиназой. Первая содержится в печени, осуществляющей фосфорилирование основной массы глюкозы, тогда как многочисленные гексокиназы распределены по тканям более широко. Глюкозо-6-фосфат (Г-6-Ф) превращается в глюкозо-1-фосфат (Г-1-Ф) в обратимой реакции, катализируемой фосфоглюкомутазой. Уридиндифосфатглюкоза (УДФГ) синтезируется из Г-1-Ф и уридинтрифосфата под действием УДФГ-пирофосфорилазы. Генетическая недостаточность ни одного из этих печеночных ферментов не была зарегистрирована. Молекула гликогена затем удлиняется путем присоединения отдельных остатков глюкозы из УДФГ, в результате чего образуется полимер. Эта реакция катализируется гликогенсинтазой, которая существует в активной дефосфорилированной и неактивной фосфорилированной формах. Для синтеза нормально разветвленной молекулы гликогена требуется также участие ветвящего (бранчер) фермента (1,4-a-гликан: 1,4-a-гликан-6-гликозилтрансфераза), который пе-реводит 1,4-связи олигосахарида в положение 1,6-связей.
Глюкоза мобилизуется из гликогена целым рядом ферментативных реакций. На гликоген непосредственно действует активная форма фосфорилазы — фосфорилаза а, отщепляя отдельные остатки глюкозы и образуя Г-1-Ф. В печени и мышцах фосфорилаза кодируется разными генными продуктами. В этих тканях фермент может существовать в активной фосфорилированной и неактивной дефосфорилированной формах. Фосфорилаза представляет собой димер, состоящий из одинаковых субъединиц, причем обе формы фермента подвергаются сложной аллостерической регуляции. Неактивная фосфорилаза b превращается в активную форму под действием фосфорилазо-b-киназы, которая существует и в активной фосфорилированной и неактивной дефосфорилированной формах. Этот фермент состоит из четырех разных субъединиц (a, b, g, d4),причем d-цепь идентична связывающему кальций белку — кальмодулину. Скорость мобилизации глюкозы этой системой регулируется каскадом киназных реакций, включающих цАМФ.
По характеру ферментной недостаточности принято различать 12 типов гликогенозов, среди которых выделяют печеночные (гликогенозы 0, I, III, IV, VI, VIII, IX, Х, ХI типов) или мышечные формы (гликогенозы V и VII типов). Гликогеноз II типа проявляется поражением только мышц или поражением многих систем и органов (генерализованная форма). Также возможны сочетания нескольких типов.
Клиническая картина гликогенозов характеризуется гипогликемией (рвота, судороги, потеря сознания, кома). Течение болезни зависит от места депонирования гликогена: печень, почки, мышечная ткань. Соответственно выделяют цирроз печени, почечную форму, мышечный синдром (включая сердечную форму). Преобладание у новорожденного ребенка симптомов гипогликемии может привести к синдрому внезапной смерти. Прогноз зависит от типа болезни.
Классификация основана на различиях в дефектах ферментов, лежащих в основе заболеваний.
Гликогеноз 0 типа (агликеноз) характеризуется резким снижением запасов гликогена в печени, наблюдается тяжелое состояние вплоть до развития комы (гипогликемический синдром). Кома может возникать после рождения при позднем прикладывании ребенка к материнской груди, а позднее — утром натощак и в перерывах между кормлениями. При отсутствии лечения ребенка наступает нарушение психомоторного развития.
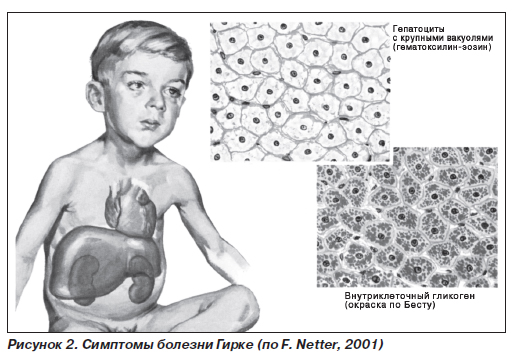
Гликогеноз I типа (болезнь Гирке) (рис. 2) — гликогеноз, обусловленный недостаточностью глюкозо-
Гликогеноз II типа (болезнь Помпе) — наследуется по аутосомно-рецессивному типу. Симптомы проявляются в первые недели жизни — до шести месяцев после рождения. Дефект фермента найден в печени, почках, селезенке, мышцах, нервной ткани, лейкоцитах. Наблюдается расстройство дыхания, беспокойство или адинамия. Отмечаются отсутствие аппетита, задержка роста, мышечная гипотония. Увеличиваются размеры сердца, печени, почек, селезенки. Сердце приобретает шаровидную форму, в связи с гипертрофией миокарда появляются изменения ЭКГ. Часто возникают гипостатические пневмонии, бронхиты, ателектазы легких, наблюдаются миодистрофия, гипорефлексия, спастические параличи. Мышечная форма гликогеноза II типа возникает только в мышцах при дефиците кислой α-1,4-глюкозидазы. Болезнь проявляется в более поздние сроки и по клинической картине напоминает миопатию.
Гликогеноз III типа (болезнь Кори, болезнь Форбса, лимитдекстриноз) — гликогеноз, вызванный недостаточностью фермента амило-1,6-глюкозидазы. Этот фермент катализирует расщепление связей […C-O-C…] в молекуле гликогена в точках ветвления. Болезнь сопровождается отложением атипичного гликогена в печени, сердце, мышцах. Наследуется по аутосомно-рецессивному типу. Дефект фермента найден в печени, мышцах, лейкоцитах, эритроцитах. С первых месяцев жизни ребенка наблюдаются гепатомегалия, мышечная гипотония, гипертрофия отдельных групп мышц. В некоторых случаях у больных отмечаются нарушение сердечной проводимости и кровообращения, гипертрофия миокарда. Развитие заболевания замедляется после пятилетнего возраста или в пубертатном периоде.
Гликогеноз IV типа (болезнь Андерсена, амилопектиноз, диффузный гликогеноз с циррозом печени) — гликогеноз, семейный цирроз печени, вызванный дефектом фермента амило-(1,4-1,6)-трансглюкозидазы. Этот фермент катализирует превращение 1,4-связей в молекуле гликогена в 1,6-связи, то есть обусловливает ветвление молекулы полисахарида. Заболевание сопровождается избыточным накоплением атипичного гликогена в печени. Наследуется по аутосомно-рецессивному или связанному с полом типу. Дефект фермента найден в печени, почках, мышцах, лейкоцитах. Болезнь наблюдается с первых месяцев жизни и характеризуется гепатоспленомегалией, развитием цирроза печени, желтухой, гипогликемией.
Гликогеноз V типа (болезнь Мак-Ардла, миофосфорилазная недостаточность) — гликогеноз, связанный с дефектом мышечной фосфорилазы. Заболевание, обусловленное нарушением каталитической функции этого фермента; сопровождается отложением значительного количества гликогена в мышцах. Наследуется по аутосомно-рецессивному типу. Дефект фермента найден в мышцах. В связи с гликогенной инфильтрацией скелетные мышцы увеличиваются в объеме, становятся очень плотными. Мышечная слабость, мышечные спазмы, тахикардия при физической нагрузке появляются в первые десять лет жизни и прогрессируют. Наблюдается транзиторная миоглобинурия. Концентрация лактата в крови после физической нагрузки уменьшается. Чаще (в 5 раз) болеют лица мужского пола.
Гликогеноз VI типа (болезнь Герса, гепатофосфорилазная недостаточность) — гликогеноз, вызванный недостаточностью фосфорилазы печени. Фосфорилаза печени катализирует фосфорилирование гликогена с образованием глюкозо-1-фосфата. Нарушение этого механизма приводит к избыточному отложению гликогена в печени. Наследуется, предположительно, по аутосомно-рецессивному типу. Проявляется обычно на первом году жизни. Характерны значительное увеличение печени в результате гликогенной инфильтрации гепатоцитов, задержка роста, кукольное лицо, гиперлипидемия, гипергликемия после внутривенного введения галактозы, повышенное содержание гликогена в эритроцитах. Наследуется по аутосомно-рецессивному типу. Дефект фермента найден в печени, лейкоцитах. Проявляется обычно на первом году жизни.
Гликогеноз VII типа (болезнь Таруи, миофосфофруктокиназная недостаточность) — симптомы сходны с гликогенозом V типа. Дефект фермента найден в мышцах, эритроцитах. Также характерны мышечная слабость, утомляемость и отсутствие гиперлактацидемии после физической нагрузки.
Гликогеноз VIII типа (болезнь Томсона) — встречается редко, наследование не установлено. Дефект фермента найден в печени, в головном мозге. После рождения увеличиваются размеры печени, появляются нистагм («танцующие глаза») и атаксия. Неврологическая симптоматика прогрессирует.
Гликогеноз IX типа (болезнь Хага) — наследуется по рецессивному, связанному с полом типу. Дефект фермента найден в печени. У больных наблюдается гепатомегалия.
Гликогеноз Х типа — известен случай у единственного больного, наследование не установлено. Дефект фермента найден в печени, мышцах. Наблюдалась гепатомегалия, через 6 лет после начала заболевания появились мышечные боли и спазмы мышц после физических упражнений.
Гликогеноз XI типа (болезнь Фанкони — Бикеля) — наследование не установлено. Дефект фермента найден в печени, почках. Характеризуется значительным увеличением печени и резкой задержкой роста. Наблюдаются симптомы гипофосфатемического рахита. В пубертатном периоде возможны уменьшение размеров печени, ускорение роста, нормализация уровня фосфора в крови.
Лечение гликогенозов в основном симптоматическое и направлено на изменение нарушений обменных процессов. Цель лечения — предупредить тяжелую гипогликемию. Назначают диету, богатую белками и углеводами. Питание должно быть частым (каждые 4 ч). Белки служат источником аминокислот — субстратов глюконеогенеза; они уменьшают углеводную нагрузку, приводящую к гипергликемии и лактат-ацидозу. Такая диета предотвращает гипогликемию и кетоацидоз натощак, уменьшает гипергликемию и лактат-ацидоз после еды и способствует ускорению роста. При мышечных формах гликогенозов улучшение отмечается при соблюдении диеты с высоким содержанием белка, назначении фруктозы, поливитаминов, АТФ. Иногда необходимо применение глюкагона, анаболитических гормонов, глюкокортикоидов. Предпринимаются попытки введения больным недостающих ферментов.
Профилактика не разработана.
Мукополисахаридозы
Этиология и патогенез. Мукополисахаридозы (МПС) — гетерогенная группа заболеваний, отнесенных к наследственным болезням обмена сложных сахаров. МПС сопровождаются избыточным накоплением в тканях и повышенной экскрецией гликоз-аминогликанов (ГАГ) — кислых мукополисахаридов, соединенных с белком и состоящих из уроновых кислот, аминосахароз и нейтральных сахаров. Указанные комплексы существуют в форме протеогликанов, являющихся важнейшими компонентами основного структурного белка волос (0-кератина) и структурного белка соединительной ткани (коллагена).
Для большинства МПС характерен аутосомно-рецессивный тип наследования, кроме синдрома Хантера (Х-сцепленный рецессивный).
Клиническая картина. Манифестация болезни, как правило, в возрасте до 7 лет, задержка роста (до карликовости), контрактура суставов, кифоз/кифосколиоз/сколиоз, массивный череп с глубоким и удлиненным турецким седлом, короткая шея, деформация грудной клетки, веслообразные ребра, укорочение трубчатых костей, грубые черты лица, помутнение роговицы, гепатоспленомегалия, задержка психического развития, судороги, глухота, грыжи (пупочная, паховая, пахово-мошоночная), врожденные пороки сердца, гликозаминогликанурия (100–200 мг в сутки). Характерные для МПС признаки дисморфизма получили название «гаргоилический фенотип» (гаргоилизм — синоним МПС).
Среди МПС выделяют ряд типов, каждый из которых обусловлен дефицитом специфической лизосомной гидролазы, участвующей в последовательном расщеплении ГАГ.
I тип — синдром Гурлера (4р16 — IDUA). Выделяют подтипы: Гурлер, Шейе, смешанный вариант. Для всех характерно снижение активности альфа-идуронидазы и накопление в тканях дерматан- и гепарансульфатов.
II тип — синдром Хантера (Гюнтера) (Хq28 — IDS). Снижение активности L-идуросульфат-сульфатазы и отложение в тканях дерматан- и гепарансульфатов. Клинические признаки менее выражены, продолжительность жизни больше, чем при других типах МПС.
III тип — синдром Санфилиппо (12q13.4). В зависимости от природы первичного биохимического дефекта выделяют четыре подтипа: А, В, С, D. В клинической картине преобладают психические расстройства: деменция, агрессивность. Продолжительность жизни не превышает 20 лет.
IV тип — синдром Моркио (16q24.3 — GALNS). Выделяют подтипы А и В. В тканях откладывается кератансульфат. Преобладают поражения скелета и непропорционально низкий рост.
V тип — синдром Шейе (см. I тип).
VI тип — синдром Марото — Лами (5q11.2 — ARSB). Дефицит фермента арилсульфатазы В. В тканях накапливается дерматансульфат. Фенотипически напоминает МПС I типа, но интеллект не снижен.
VII тип — синдром Слая (7q21.11 — GUSB). Дефицит фермента δ-глюкуронидазы. В тканях накапливаются дерматан-, гепаран- и хондроитин-сульфаты. Фенотипиче-ски напоминает МПС I типа, но имеет более доброкачественное течение.
Диагностика МПС основывается на совокупности данных генеалогического анализа, клинических проявлений, типичных рентгенологических данных, экскреции с мочой оксипролина (снижение), ГАГ и их фракций (превышение в 5–10 раз). Точная идентификация типов МПС возможна только с помощью определения активности лизосомных гидролаз в лимфоцитах и лейкоцитах крови, культуре фибробластов кожи, биоптатов печени, а также в моче.
Лечение больных МПС в основном симптоматическое, заключается в назначении терапии, способствующей нормализации или стабилизации патологического процесса в опорно-двигательном аппарате, сердечно-сосудистой и центральной нервной системах, паренхиматозных органах, органах зрения и слуха. Перспективным считается плазмаферез.