
Газета «Новости медицины и фармации» Гастроэнтерология (304) 2009 (тематический номер)
Вернуться к номеру
Недостаточность альфа-1-антитрипсина
Авторы: Н.Е. Моногарова, Т.В. Мороз, А.А. Минаев, Донецкий национальный медицинский университет им. М. Горького, Кафедра внутренней медицины им. А.Я. Губергрица
Версия для печати
Одним из заболеваний, требующих молекулярной диагностики, является альфа-1-антитрипсиновая недостаточность (ААТН) — распространенная наследственная болезнь, обусловленная сниженной концентрацией альфа-1-антитрипсина (А1-АТ) в сыворотке крови вследствие различных мутаций в гене Pi, проявляющаяся чаще всего в виде хронических неспецифических заболеваний легких с развитием эмфиземы, а также поражением печени и сосудов.
Что такое альфа-1-антитрипсин
Это гликопротеид, который синтезируется в печени (рис 1.). Альфа-1-антитрипсин тормозит действие трипсина, химотрипсина, эластазы, калликреина, катепсинов и других ферментов тканевых протеаз. Дефицит А1-АТ приводит к повышенному накоплению протеолитических энзимов и последующему повреждению тканей. Однако известно, что при дефиците А1-АТ поражения легких и печени не всегда бывают тяжелыми и необратимыми. Видимо, данный дефицит может быть компенсирован другими механизмами.
У некоторых больных в гепатоцитах обнаруживают ШИК-позитивные шары. Примерно у 10 % детей, гомозиготных по аллелю Z, отмечается тяжелое поражение печени, включая гепатит у новорожденных и прогрессирующий цирроз печени. Полагают, что 15–20 % хронических гепатитов у грудных детей обусловлены ААТН. У взрослых ААТН чаще всего приводит к мелкоузловому циррозу печени, который протекает бессимтомно, со временем может перейти в крупноузловой цирроз печени, иногда развивается печеночноклеточный рак.
Частота поражения печени не зависит от частоты поражения легких.
Эпидемиология ААТН
Предположительно 60 000–100 000 американцев имеют ААТН. Установлено, что из 14 млн американцев с хроническими неспецифическими обструктивными заболеваниями легких 2 млн имеют эмфизему. В группе из 965 пациентов с эмфиземой выявлено, что частота тяжелой ААТН составляет 2–3 %. Сопоставив эти цифры, можно прогнозировать, что примерно 63 000 американцев страдают эмфиземой, обусловленной дефицитом А1-АТ. Многие исследования показывают, что в настоящее время диагностирован лишь небольшой процент от общего числа больных с ААТН — всего 4–4,5 %.
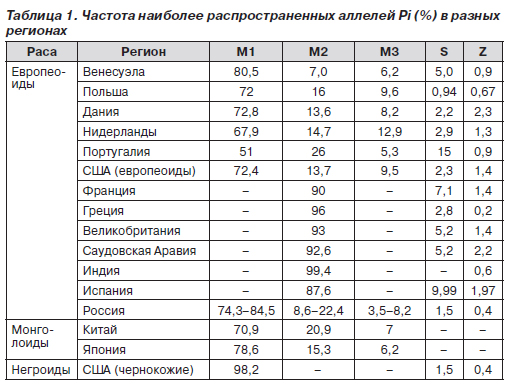
В табл. 1 приведена частота основных аллелей гена Pi в некоторых регионах мира.
Структура гена и номенклатура аллелей
Ген Pi, ответственный за синтез А1-АТ, картирован на длинном плече хромосомы 14. Известна его полная нуклеотидная последовательность размером 12 kb. Ген имеет пять экзонов, причем экзон I подразделяется на Ia, Ib и Iс. Экзоны Iа и Ib содержат фрагменты, ответственные за транскрипцию в макрофагах, а экзон Iс имеет промоторную область, необходимую для транскрипции в гепатоцитах. Кодирующая область гена захватывает четыре экзона (II–V). Старт-кодон для 24-аминокислотного сигнального пептида и два из трех сайтов гликозилирования белковой молекулы локализованы в экзоне II. В экзоне III располагается третий сайт гликозилирования и наиболее полиморфный сайт в кодирующей области Val213Ala. В экзоне V расположен активный сайт М358. В этом экзоне наиболее часто встречаются мутации, приводящие к ААТН (Z-вариант).
Номенклатура аллелей гена Pi основана на электрофоретической подвижности продуктов этих аллелей. Варианты А1-АТ, двигающиеся наиболее быстро к аноду, названы первыми буквами латинского алфавита. Многочисленные аллели гена можно подразделить на нормальные, дефицитные, нулевые и аллели с измененными свойствами. Наиболее частый дефицитный вариант — аллель Z — двигается медленно и расположен очень близко к катоду, в Z-области. Нормальные аллели обычно продвигаются к середине геля и попадают в М-область, поэтому называются чаще всего М-аллелями.
Патогенез и нозологические формы ААТН
А1-АТ (серпин) (serpin — serine protease inhibitor — сывороточный ингибитор протеаз) является одним из представителей семейства сериновых протеаз, к которым относят также антитромбин, контролирующий свертывание крови; С-ингибитор, регулирующий реакции каскада системы комплемента; различные ингибиторы плазминогена, останавливающие процесс фибринолиза.
Серпин представляет собой гликопротеин с молекулярной массой 52 кД и размером в 418 аминокислот. Синтез его проходит главным образом в печени и в меньших количествах – в мононуклеарных фагоцитах и нейтрофилах. Главной функцией белка является инактивация различных групп протеаз, секретируемых лейкоцитами при реакциях неспецифической защиты организма. При воспалении уровень А1-АТ может возрастать в три раза, вследствие чего его относят к маркерам острофазового воспаления.
Структура белка представляет собой совокупность бета-слоев и редких альфа-спиралей, имеющих четыре боковые углеводные цепи, одна из которых представлена сиаловой кислотой. Такое строение позволяет быстро изменять конфигурацию молекулы с формированием комплекса серпин-эластаза или комплекса с другой протеазой.
Прежде чем рассмотреть патогенез поражения легких и других органов при ААТН, необходимо отметить, что А1-АТ является основной антипротеазой, которая нейтрализует избыток протеаз, продуцируемых как микроорганизмами, так и клетками макроорганизма. А1-АТ синтезируется в печени в шероховатой эндоплазматической сети. Он содержится в альфа-1-фракции белков сыворотки крови и составляет 80–90 % всех альфа-1-глобулинов сыворотки. А1-АТ ингибирует трипсин и другие протеазы. А1-АТ является основным ингибитором эластаз, выделяемых альвеолярными макрофагами и полиморфноядерными лейкоцитами, обеспечивая 90 % антиэластазной активности (10 % приходится на альфа-2-макроглобулин, альфа-антихимотрипсин и низкомолекулярные тканевые эластазы). Имея сравнительно небольшую молекулярную массу (54 000), A1-AT хорошо проникает в ткани; он выполняет и транспортную функцию, возвращаясь вместе со связанной протеазой в кровяное русло, где она подвергается действию других ингибиторов и ретикулоэндотелиальной системы.
Патология легких
Патогенетические механизмы развития заболевания различных органов неодинаковы. В отношении патогенеза поражения легких наиболее признанной является теория нарушения протеазно-антипротеазного равновесия (рис. 2–5).
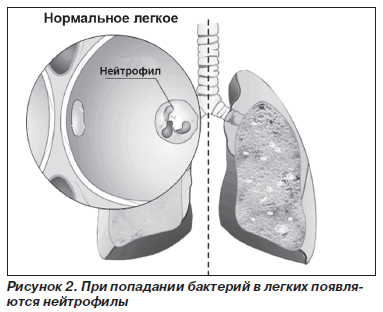
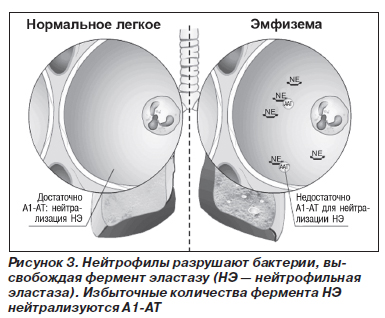
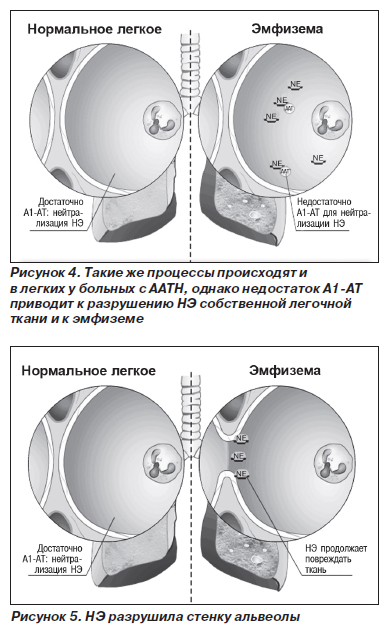
В норме миллимолярная концентрация лейкоцитарной эластазы из азурофильных гранул нейтрофилов создает кратковременный взрыв протеолитической активности до момента подавления реакции перицеллюлярными ингибиторами (антипротеазами). Продолжительность воздействия агрессивных ферментов на легочную ткань не превышает в норме 20 миллисекунд. В результате снижения концентрации А1-АТ в крови время контакта ферментов с тканью легких может удлиняться до 80 миллисекунд, что приводит к неизбежной деструкции эластических волокон легких. Механизм развития эмфиземы легких при ААТН неизвестен. Установлено, что А1-АТ подавляет активность трипсина, эластазы и некоторых других протеаз. Эксперименты показали, что он защищает легочную ткань от протеаз, высвобождаемых лейкоцитами, и тем самым сохраняет структурную целостность эластина. Можно предположить, что к эмфиземе легких приводит хроническое воспаление (вследствие инфекции или загрязнения воздуха), поскольку при ААТН легочная ткань ничем не защищена от протеаз лейкоцитов, привлеченных в воспалительный очаг.
Протеазам лейкоцитов принадлежит ведущая роль в патогенезе эмфиземы легких не только у больных с ААТН. Имеется немало данных, что высвобождаемые нейтрофилами и альвеолярными макрофагами протеазы могут вызвать эмфизему легких даже при нормальном содержании ингибиторов протеаз в крови. Причина, возможно, кроется в том, что концентрация протеаз в тканях выше, чем концентрация ингибиторов протеаз, либо некоторые протеазы нечувствительны к ингибиторам или недоступны для них. От того, какая из указанных причин играет основную роль в патогенезе эмфиземы легких, будет зависеть судьба разрабатываемых в настоящее время препаратов — экзогенных ингибиторов протеаз.
Замещаясь соединительной тканью, паренхима легких со временем теряет свою эластичность; развиваются обструктивные явления; формируется эмфизема, которая возникает первично, на фоне хронического бронхита или другого хронического неспецифического заболевания легких. Чаще всего расширяется весь ацинус, и эмфизема характеризуется как панацинарная. Буллезные изменения наиболее выражены в основании легких, а не на верхушках, что больше характерно для эмфиземы, не связанной с ААТН. Среди всех гомозигот по PiZ около 85 % имеют рентгенологические признаки эмфиземы; из них практически в 100 % случаев отмечаются эмфизематозные изменения в базальных отделах легких (рис. 6).
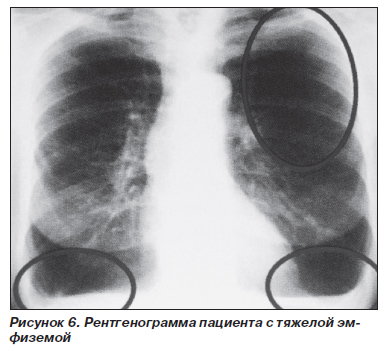
Риск развития эмфиземы значительно возрастает при снижении уровня сывороточного А1-АТ менее 0,8 г/л (11 ммоль/л) (норма — 2,0–4,0 г/л). Как правило, в клинике у таких пациентов отмечается одышка (67–98 %), которая значительно снижает качество жизни пациентов и заставляет их впервые обратиться к врачу. Кроме эмфиземы ААТН может проявляться идиопатическим фиброзом, бронхоэктазами; имеются данные о ее связи с развитием рака легких.
Некоторые аллели гена муковисцидоза (CF) способствуют развитию диссеминированных бронхоэктазов. Описаны случаи сочетания муковисцидоза и ААТН, причем одни аллели гена CF предрасполагают к более доброкачественному течению процесса, другие — к более тяжелому. Среди больных асбестозом отмечается возрастание частоты аллеля PiS в 4 раза, что свидетельствует о предрасположенности пациентов с ААТН к развитию данной профессиональной патологии.
Патология печени
На сегодняшний день принято считать, что одной из главных причин поражения печени является агрегация плохо растворимого белка PiZ в эндоплазматическом ретикулуме гепатоцитов. Примерно до 85 % синтезированного белка PiZ неспособно покинуть гепатоциты, и скопления дефектного протеина можно обнаружить в виде кислых включений, окрашивающихся по Шиффу. Скорость аккумуляции патологического продукта гена зависит от двух факторов: скорости синтеза белка и температуры тела. Полимеризация Z-продукта очень быстро происходит при температуре тела 41 °С. Пациентам, гомозиготным по аллелю Z, склонным к выраженному гипертермическому ответу уже при легких простудах, показано экстренное снижение температуры тела даже при субнормальных значениях гипертермии.
У небольшого числа людей, имеющих ААТН, развивается цирроз печени уже в младенческом и раннем детском возрасте. Из всех новорожденных и младенцев, гомозиготных по генотипу PiZ, явные клинические проявления гепатита и цирроза могут быть обнаружены у 10 %. Факторами риска развития печеночной симптоматики в детстве являются затяжная гипербилирубинемия в первые недели жизни, мужской пол и инфицирование вирусом гепатита В. Фактором, снижающим риск, является кормление грудью. Примерно 10 % пациентов с клиническими симптомами в младенчестве погибают к восьми годам. В странах с развитой трансплантационной хирургией ААТН, наряду с атрезией желчевыводящих протоков, является ведущим показанием к трансплантации печени у детей.
В последнее время показано, что пациенты с ААТН склонны к инфицированию вирусом гепатита С. При исследовании групп больных с тяжелыми болезнями печени выявлено, что среди различных патологий отмечается особенно высокая гетерозиготность PiMZ в группах больных с вирусными гепатитами В и С, алкогольным циррозом печени, первичными гепатокарциномами, криптогенным циррозом печени и рядом других болезней.
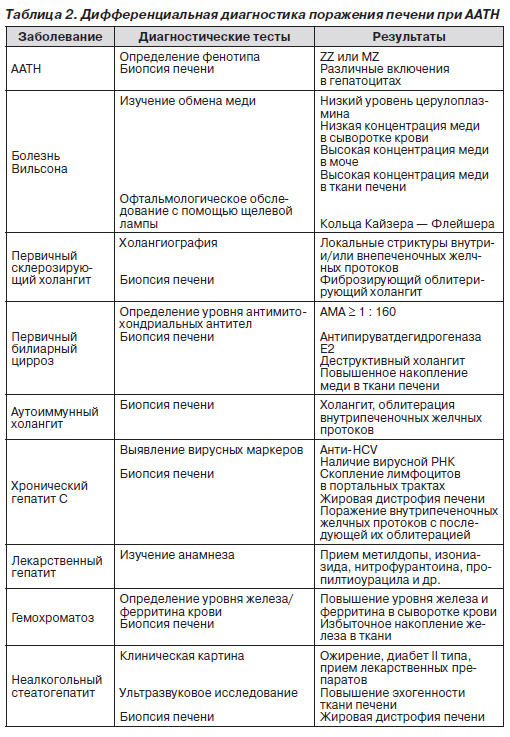
Биопсия печени. Морфологические изменения печени зависят от стадии заболевания. У новорожденных отмечаются следующие изменения: гигантские гепатоциты, холестаз, умеренный стеатоз, портальный фиброз, пролиферация желчных протоков. При дальнейшем прогрессировании заболевания развивается выраженный портальный фиброз и цирроз печени. Характерными признаками ААТН является агрегация эозинофилов и наличие PAS-положительных диастазорезистентных глобулярных включений в эндоплазматическом ретикулуме перипортальных гепатоцитов. Основные критерии дифференциальной диагностики поражения печени при ААТН представлены в табл. 2.
Клиника
Дефицит А1-АТ приводит к поражению нескольких органов и систем: заболеваниям печени у новорожденных, детей и взрослых, ранней эмфиземе легких у взрослых, мембранозно-пролиферативному гломерулонефриту, фиброзу поджелудочной железы.
Поражение печени регистрируется при фенотипах PiZZ и PiMZ. У 20 % новорожденных, имеющих фенотип PiZZ, развивается неонатальный холестаз, а желтуха служит первым симптомом недостаточности А1-АТ. Гепатомегалия обычно умеренная, может наблюдаться спленомегалия.
В анализах крови выявляются прямая гипербилирубинемия (увеличение в 4–20 раз), выраженное повышение активности маркеров холестаза: гамма-глутамилтранспептидазы (ГГТП) и щелочной фосфатазы (ЩФ), умеренное увеличение трансаминаз — аланиновой (АлАТ) и аспарагиновой (АсАТ).
Исход неонатального холестаза вариабелен. В большинстве случаев он самостоятельно разрешается к 3–4-му месяцу, в некоторых случаях заболевание может прогрессировать с развитием печеночной недостаточности к 6–8-му месяцу жизни ребенка.
Сохраняющаяся гепатомегалия и повышенные показатели трансаминаз у больных с разрешившимся холестазом рассматриваются как неблагоприятные факторы и предсказывают формирование цирроза печени в среднем в течение 6 лет. Если печеночные энзимы возвращаются к норме и нет гепатомегалии, то прогноз лучше. Общий риск развития цирроза и смерти от заболевания печени у больных с неонатальным холестазом составляет около 60 %. У 20 % больных происходит самостоятельное разрешение заболевания печени.
Риск развития хронического гепатита и цирроза у взрослых с фенотипом PiZZ увеличивается с возрастом и составляет 2 % в возрасте 20–40 лет, 5 % — в возрасте 40–50 лет и 15 % — в возрасте старше 50 лет. Риск развития гепатоцеллюлярной карциномы (ГЦК) у больных этих групп составляет 2–3 %. Поражение легких в виде раннего развития эмфиземы диагностируется в молодом возрасте. Пациенты отмечают клинику хронического обструктивного заболевания легких: кашель, одышку, обструктивный тип нарушения функции дыхания.
Лечение
Лечение ААТН состоит из нескольких этапов и включает в себя:
1. Первичную профилактику (прекращение курения, адекватное питание и физические упражнения).
2. Лечение сопутствующей патологии (бронхиальная астма, профилактика и своевременное лечение инфекций дыхательных путей и легких, гепатитов).
3. Специфическую терапию. Специфическое лечение основывается на заместительной аугментационной (от англ. augmentation — приращение, увеличение) терапии экзогенным А1-АТ для нормализации протеазно-антипротеазного равновесия в сыворотке крови и базальных отделах легких.
Предложены следующие критерии для рассмотрения возможности экзогенной заместительной терапии:
Поскольку в современной литературе нет описания случаев развития преждевременной эмфиземы при уровне А1-АТ 0,8 г/л (11 ммоль/л) и выше, то именно концентрация ниже этого порога была выбрана как показание для заместительной терапии. Наиболее широко распространенный препарат данной группы — проластин, получаемый из сыворотки крови и представляющий концентрат А1-АТ. В настоящее время около 2200 человек получают проластин в США; он разрешен к применению в Канаде, Германии, Испании и ряде других стран, где его получают еще около 2000 человек. Проластин практически не вызывает аллергических и анафилактических реакций. Стоимость внутривенной инъекции проластина (из расчета 60 мг/кг массы тела, 1 раз в неделю) составляет от 34,6 до 67,4 доллара в зависимости от массы тела пациента. В год стоимость заместительной терапии составляет от 1660,8 до 3325,2 долларов. Побочные эффекты заместительной терапии относительно редки и проявляются слабо выраженной дыхательной недостаточностью. Возможно ухудшение состояния на фоне лечения в связи с перегрузкой белком. В настоящее время разрабатываются препараты, которые можно было бы применять ингаляционно.
В некоторых случаях возможна стимуляция выработки эндогенного А1-АТ. При этом подходе к терапии пациенты принимают лекарства, которые стимулируют синтез и секрецию А1-АТ из гепатоцитов. К таким препаратам относятся даназол, тамоксифен и эстроген-прогестиновые препараты. Подобную терапию могут получать пациенты с мягкими фенотипами, например SZ. Препарат дапсон также относится к этой группе и применяется для лечения панникулита.
Примерно 12 % всех трансплантаций легких выполняются по поводу эмфиземы, обусловленной ААТН. Пятилетняя выживаемость после данной манипуляции составляет 45 %. Такая операция, как редукция объема легочной ткани, представляет собой иссечение наиболее пораженных эмфиземой участков легочной ткани, определенных при помощи визуализирующих методик. Смертность после этой операции невелика и составляет около 5 %. Положительные эффекты операции сохраняются в течение одного года. При прогрессировании поражений печени единственным методом лечения является трансплантация печени или групп гепатоцитов.
Типы скринингов на ААТН
К типам скринингов на ААТН относятся:
Популяционные скрининги взрослых предпринимались среди доноров крови в США в конце 80-х годов. Первоначально для детекции нарушений секреции А1-АТ использовали полуколичественные биохимические тесты, которые позволяли определить только уровень ААТ в крови, причем с большим процентом ложноотрицательных результатов. Данный метод использовал S. Eriksson (1962), открывший наследственную ААТН. В связи с появлением метода изоэлектрического фокусирования (ИЭФ) открылись новые возможности в определении гена Pi.
Скрининг с использованием ИЭФ выявил большое количество недиагностированных пациентов. С
Неонатальные скрининги проводятся спорадически в разных странах. Самые большие программы скринирования были осуществлены в 70-е годы в Швеции и США (Орегон) (200 000 и 107 038 новорожденных соответственно). Использовался метод ИЭФ и иммунный лидазный тест. Среди новорожденных в США частота аллеля Z встречается в одном случае на 5097 детей; в Швеции этот показатель значительно выше и составляет один случай на 1575 новорожденных. Такие исследования наиболее достоверно показывают отягощенность по ААТН населения данных регионов. Эти программы позволили осуществить длительное наблюдение и изучение течения заболевания у детей и взрослых. В наши дни пилотные исследования новорожденных проводятся в США, Чехии, Бельгии и других странах.
Опыт скринирования 18-летних подростков в Европе показывает, что этот возраст слишком поздний для выявления ААТН, так как значительная часть больных уже начала курить и у них уже проявляются симптомы поражения легких. Во Франции скрининг на ААТН проводится наряду со скринингом на фенилкетонурию, врожденный гипотиреоз, муковисцидоз, адреногиперплазию и недостаточность биотинидазы. Считается, что выявление именно этих заболеваний, их раннее лечение и профилактика выгоднее, чем социальное обеспечение больных, их лечение при выставлении диагноза в позднем возрасте. Справедливость этого положения подтверждают показатели смертности. В США смертность от ААТН за период с 1979 по 1991 гг. превысила 32 миллиона человек, среди которых 528 115 новорожденных и 219 557 детей до 14 лет.
Национальные скрининговые программы
Национальные ассоциации и общества больных с ААТН функционируют в США (с
Несмотря на довольно широкую распространенность ААТН и несомненные преимущества ее ранней диагностики, лишь в десятке стран (Франция, США, Испания и т.д.) осуществляются скрининговые программы по выявлению этой патологии. Для этой цели в основном используют модель Laurel с незначительными модификациями.
В Швеции проводили несколько пилотных скрининговых программ исследования населения разных возрастов, используя для определения ААТН качественный метод изоэлектрического иммунного фокусирования в агарозном геле с антителами к А1-АТ и трансферрину. Данный метод обладает недостаточной точностью: его чувствительность — 83,6 %, а специфичность — 61,4 %. Необходимость исследования сыворотки требует забора от 5 мл крови, что довольно много для новорожденного. Эпизодичность исследований не позволяет рассматривать данную модель как основу для построения постоянно действующей схемы скринирования населения. В настоящее время в Швеции прошла апробацию и активно внедряется в практику американская модель скрининга.
В некоторых странах с целью диагностики используется иммуноферментный лидазный метод, который может давать ложноотрицательные результаты, т.к. система А1-АТ реагирует на функциональное состояние организма, а чувствительность данной методики не намного выше, чем метода изоэлектрического иммунного фокусирования (87 и 68 % соответственно).
В США используют практику пилотных исследований новорожденных с предоставлением всем желающим возможности прохождения этой платной процедуры. Основной упор делается на платное обследование с предоставлением заинтересованным лицам самого широкого спектра информации по данной патологии и на высокую грамотность специалистов, направляющих на обследование. Для генотипирования наиболее часто используют рестрикционный анализ и секвенирование V экзона. Активно осуществляются программы по социальной адаптации больных и носителей патологических аллелей.
Используемая практика также не лишена недостатков, так как нерегулярное проведение исследования новорожденных снижает эффективность скрининга и увеличивает количество несвоевременно диагностированных случаев. Кроме того, упор на платные обследования приводит к высокому вкладу социальных аспектов в достоверность и охват скрининга. Сюда же следует отнести проблемы со стороны пациентов, которые могут недооценить возможные последствия от несвоевременной диагностики патологического состояния.
В настоящее время ведутся активные разработки биочипов для детекции дефицитных аллелей гена Pi.
Алгоритмы скрининга для каждой группы разрабатываются индивидуально.
Целесообразность исследования новорожденных на дефицитные варианты гена Pi определяется возможностью ранней диагностики и доклинического ведения носителя, не допуская трансформации патологического генотипа в фенотип под воздействием факторов среды.
Ввиду обширности этих групп для скрининга нужно рассматривать нозологические единицы, вклад в патогенез которых гена Pi описан в литературе: для поражения бронхолегочной системы — эмфизема, бронхиальная астма, хронический бронхит; для поражения гепатобилиарной системы — ювенильный цирроз, гепатиты (в том числе вирусные), рак печени.
Исследование осуществляется методом ДНК-диагностики с использованием ПЦР и рестрикционного анализа, поскольку такая диагностика обладает рядом неоспоримых преимуществ по сравнению с другими методиками — дешевизна, простота, чувствительность (100 %) и информативность.
Выявленная группа носителей является потенциальной группой риска; для них осуществляется постановка на учет, консультации родителей, периодические осмотры, при необходимости — адекватная терапия с учетом имеющейся нозологии.
В группе риска необходимо проведение генотипирования членов семей с последующим консультированием.
Данная модель является ориентировочной, но с учетом опыта многих исследований она позволяет избежать наиболее распространенных недостатков аналогичных существующих программ.
Здоровые индивиды, не имеющие снижения ААТН или симптомов поражения органов дыхания и гепатобилиарной системы, также нуждаются в скринировании.
Также скрининг важен с точки зрения перспективы медико-генетического консультирования молодых семей в отношении прогноза рождения здорового ребенка.