
Газета «Новости медицины и фармации» Кардиология (327) 2010 (тематический номер)
Вернуться к номеру
Аритмогенна дисплазія правого шлуночка
Авторы: М.М. Долженко, Н.М. Носенко, Національна медична академія післядипломної освіти ім. П.Л. Шупика, м. Київ
Версия для печати
Аритмогенна дисплазія правого шлуночка (АДПШ) — генетичне гетерогенне спадкове захворювання серця, яке характеризується фіброзно-жировим заміщенням міокарда переважно правого шлуночка (ПШ), що супроводжується такими порушеннями ритму серця, як шлуночкова екстрасистолія (ШЕ) та правошлуночкова тахікардія (ШТ) із високим ризиком раптової серцевої смерті [5]. Вирішальний вплив на прогноз життя пацієнта з АДПШ мають злоякісні шлуночкові порушення ритму (ЗШПР).
АДПШ уперше була описана в 1977 р. Дж. Фонтейном та співавт. Дослідники звернули увагу на те, що частина хворих із резистентною до медикаментозної терапії шлуночковою тахікардією не мала інших клінічних ознак серцево-судинної патології [6]. Пізніше було описано незрозумілий зв''язок АДПШ із раптовою серцевою смертю в осіб молодого віку, які не мали ішемічної хвороби серця в анамнезі [7]. На підставі рекомендацій Всесвітньої організації охорони здоров''я АДПШ віднесена до кардіоміопатій [8].
Спадкова природа захворювання підтверджується в 30 % випадків, частіше за автосомно-рецесивним типом успадкування [9, 14]. Описано декілька сімей з цим типом успадкування АДПШ [10]. У сім''ях з аутосомно-рецесивним типом успадкування АДПШ поєднується з плантарною кератодермією та іншими дефектами сполучної тканини у вигляді «вовняного волосся» (рис. 1).
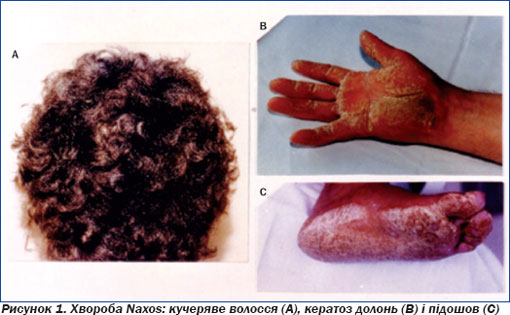
Даний фенотип захворювання називають хворобою Наксоса (уперше описано у 18 жителів грецького острова Наксос). Молекулярний аналіз виявив дефект у гені, що відповідає за плакоглобін (в одній із родин) і десмоглобін (у трьох сім''ях) [11, 12]. Плакоглобін і десмоглобін — білки, що забезпечують зв''язок десмосомальних клітин. Порушення функції десмосом може призводити до загибелі кардіоміоцитів під впливом механічного навантаження.
Гени, відповідальні за АДПШ, не до кінця ідентифіковані, однак виявлено зчеплення даного захворювання з сімома локусами, що картуються на 1, 3, 10 і 14-й хромосомах.
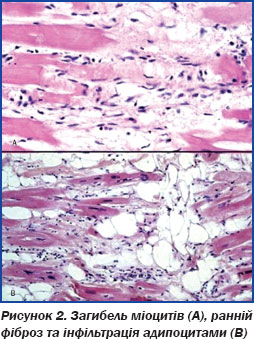
Типова гістологічна картина пр. АДПШ — загибель міоцитів (рис. 2А), ранній фіброз та інфільтрація адипоцитами (рис. 2В).
Виділяють чотири форми клінічного перебігу АДПШ [3, 13]:
— приховану (раптова серцева смерть внаслідок ШТ може бути єдиним проявом захворювання);
— аритмічну, що характеризується наявністю документованих правошлуночкових аритмій (ШЕ і ШТ) із конфігурацією комплексу QRS за типом блокади лівої ніжки пучка Гіса;
— із помірною неспецифічною суб''єктивною симптоматикою у вигляді відчуття дискомфорту в ділянці серця або неритмічності роботи серця;
— із проявами серцевої недостатності, переважно правошлуночкової.
Основний клінічний симптом АДПШ — шлуночкові аритмії з ектопічними комплексами за типом блокади лівої ніжки пучка Гіса [86]. На ЕКГ синусовий ритм може супроводжуватися змінами, що характерні для порушень процесів деполяризації й реполяризації міокарда шлуночків у правих грудних відведеннях. Також можуть бути ознаки повної чи неповної блокади правої ніжки пучка Гіса із вторинними змінами кінцевої частини шлуночкового комплексу. Найбільш специфічна ознака патології — епсилон-хвиля (рис. 3), що реєструється в початковій частині сегмента ST. Її вважають інтегральним електрокардіографічним відображенням пізніх потенціалів шлуночків, що вказує на наявність у міокарді зон із порушеним проведенням
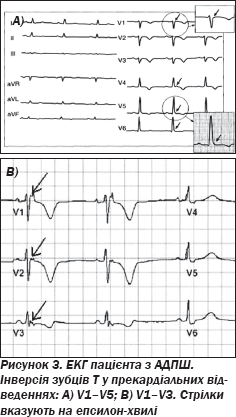
Для діагностики АДПШ використовують такі великі та малі критерії.
ЕКГ
Великий критерій:
— епсилон-хвилі або розширення комплексу QRS (> 110 мс) у правих прекардіальних відведеннях (V1–V3).
Малі критерії :
— інверсія зубця Т у правих прекардіальних відведеннях (V2 і V3) в осіб віком понад 12 років, які не мають блокади правої ніжки пучка Гіса;
— пізні потенціали шлуночків (електрокардіограма з усередненням сигналу);
— ШТ з електрокардіографічної морфологією блокади лівої ніжки пучка Гіса (стійка і нестійка) за даними ЕКГ, ХМ ЕКГ, навантажувальних проб;
— часта ШТ (> 1000/24 год) при ХМ ЕКГ.
Дані сімейного анамнезу
Великий критерій:
— спадковий характер патології, підтверджений автопсією або при операції.
Малий критерій:
— випадки раптової смерті в молодому віці (< 35 років) серед родичів, імовірно внаслідок АДПШ.
Дані ехокардіографії, магнітно-резонансної томографії й рентгеноконтрастної вентрикулографії
Великі критерії:
— значна дилатація і зниження фракції викиду ПШ без залучення ЛШ (або при незначному залученні);
— локальні аневризми ПШ (акінетичні або дискінетичні ділянки);
— значна сегментарна дилатація ПШ;
— фіброзно-жирове заміщення міокарда за даними ендоміокардіальної біопсії.
Малі критерії:
— помірна дилатація ПШ або зниження фракції викиду ПШ при нормальному ЛШ;
— помірна сегментарна дилатація ПШ;
— регіональна дискінезія ПШ.
Діагноз АДПШ можливий при виявленні двох великих, або одного великого і двох малих критеріїв [15], або чотирьох малих критеріїв.
З метою профілактики раптової серцевої смерті в разі документованих епізодів стійкої ШТ показана імплантація кардіовертера-дефібрилятора (КВДФ). При неможливості імплантації КВДФ із профілактичною антиаритмічною метою призначають антиаритмічні препарати III класу аміодарон і соталол [3, 4].
Життєво небезпечнi шлуночкові тахіаритмії при АДПШ часто є резистентними до медикаментозного лікування. Вважають, що найбільш ефективною у цих випадках є імплантація КВДФ і/або застосування амiодарону, хоча є повідомлення про добру ефективність соталолу. При АДПШ здійснюють також абляцію ектопiчних вогнищ i вентрикулотомiю.
Клінічний випадок
Пацієнтка П., 1970 року народження, звернулася на консультацію із скаргами на часті перебої в роботі серця, що виникають спонтанно, супроводжуються слабкістю, запамороченням, дискомфортом у серці.
Анамнез захворювання
Після перенесеної вірусної інфекції в січні 2002 р. уперше почали турбувати болі та дискомфорт у ділянці серця, що виникали в спокої. При обстеженні виявлено одиничні екстрасистоли. Після консультації в лікаря було призначено курс метаболічної терапії та метопролол 50 мг 2 р/день. Дискомфорт через півроку зник.
Рівно через рік, у січні 2003 р., епізод болю та перебоїв повторився, діагностовану екстрасистолію бета-блокатори не купірували. На фоні антиаритмічного лікування аміодароном порушення ритму зникли. Позитивний ефект тривав 2 роки.
У 2005 р. турбують часта шлуночкова екстрасистолія, біль та дискомфорт у серці. Аміодарон справляє позитивний ефект, проте відзначаються прояви побічної дії — фотопсія, хиткість ходи. Уперше було проведено дослідження на гормони щитоподібної залози (норма), ехокардіографія (ЕхоКГ) — патології не виявлено.
2006 р. Бета-блокатори не приносят ь полегшення при нападах аритмії. Пусковий фактор — нервове збудження. Затяжна аритмія, аміодарон в/в та pe r os за схемою. Епізод синкопе. Додаткові дослідження: гормони щи топодібної залози — у нормі, ЕхоКГ — патології не виявлено; фіброгастроезофагоскопія — грижа стравохідного отвору діафрагми, езофагіт. Призначено омепразол. Тредміл (в періоді відновлення після навантаження тригіменія), ЕхоКГ — зниження скоротливої функції правого шлуночка. Висновок: у пацієнтки парасистолія із виносного тракту правого шлуночка (ВТПШ) (А зони) до бігемінії. Рекомендована транскатетерна деструкція ектопічного вогнища — пацієнтка відмовилася.
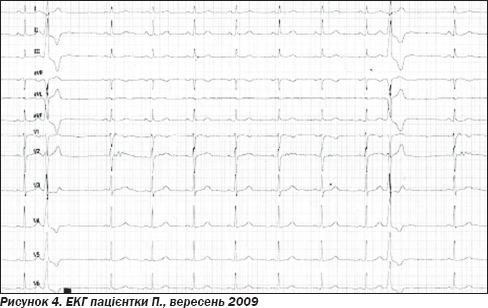
З листопада 2008 року і до цього дня: за даними холтерівського моніторування — 8–12 тисяч екстрасистол за добу. Прийом аміодарону не купірує напади аритмії. Непереносимість етацізіну — нудота, головний біль. Терапія соталолом значущого ефекту не справляє, проте відмова від соталолу призводить до збільшення кількості екстрасистол. Доза та режим терапії значення не мають (2–3 рази на день, 80–320 мг на добу). Суб''єктивно: періодично дискомфорт в ділянці серця, фізичні навантаження переносить добре, проте періодично виникає задишка при підйомі на другий-третій поверх, емоції посилюють аритмію. Гормони щитоподібної залози в нормі, гострофазові показники в нормі, ЕхоКГ — без негативної динаміки. ЕКГ — ритм синусовий неправильний, часті правошлуночкові екстрасистоли (графіка за типом блокади лівої ніжки пучка Гіса) (рис. 4).
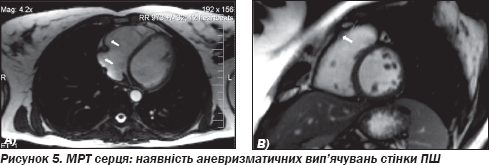
Проведено МРТ серця: виявлено патологію правого шлуночка — наявність аневризматичних вип''ячувань стінки ПШ, жирових відкладень у ВТПШ (рис. 5).
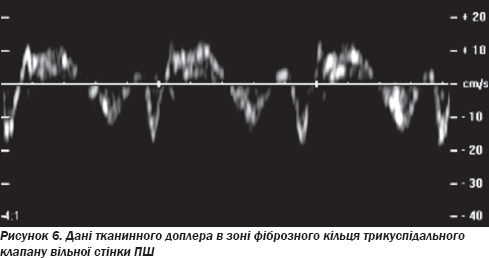
Проведено ЕхоКГ: фракція викиду лівого шлуночка та ПШ у нормі, дилатації порожнин не виявлено. При доплерівському дослідженні трансмітрального та транстрикуспідального потоків порушення діастоли відсутні. Спостерігалися невиражений фіброз модераторного тяжа та сегментарне порушення діастолічної функції в зоні фіброзного кільця трикуспідального клапана за даними тканинного доплера вільної стінки ПШ (рис. 6, 7).
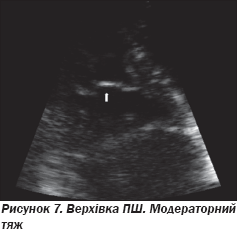
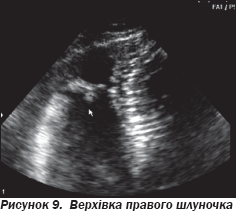
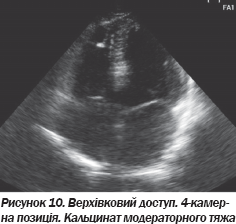
На ЕхоКГ не виявлено аневризматичних вип''ячувань ПШ, порушень його скоротливості. Незначний фіброз не можна вважати вагомим відхиленням, оскільки ці зміни не є вираженими і специфічними. На рис. 8–10 подані варіанти структурних змін модераторного тяжа в пацієнтів, які не мають патологічних аритмій.
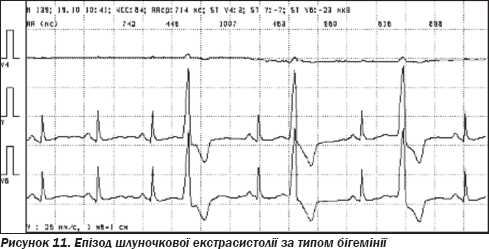
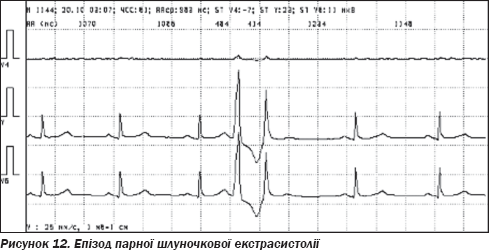
Холтерівське моніторування ЕКГ (20.10.09 на фоні лікування): на фоні синусового ритму з частотою від 44 до 112 (середня — 63) за хвилину, спостерігалася часта шлуночкова екстрасистолія, одинична, парна, періодично за типом бігемінії (рис. 11, 12). Усього 8190 (від 240 до 610, у середньому 416 на годину). Протягом дня — 6315 (480 на годину). За ніч — 1875 (319 на годину). Брадикардія в денний час. ЧСС в нічний час — у межах норми. Ішемічних зміни ST не виявлено.
Отже, у пацієнтки П., 1970 року народження, яка звернулася на консультацію зі скаргами на часті перебої в роботі серця, які виникають спонтанно, враховуючи дані обстеження, наявні два великі та один малий діагностичний критерії АДШП. Дані щодо саркоїдозу та амілоїдозу відсутні.
На даний момент хвора приймає соталол 80 мг 2 р/день (підбір дози соталолу: від 160 мг 2 р/день до 80 мг 3 р/день не був ефективним). Від інвазивних методів лікування пацієнтка поки що утрималася.
Некоронарогенні шлуночкові порушення ритму найчастіше спостерігаються в осіб молодого та середнього віку й переважно пов''язані із наслідками міокардиту, уродженими аномаліями (аритмогенна дисплазія сердця, саркоїдоз, амілоїдоз) або є ідіопатичними.
Роль провідного клінічного синдрому, що визначає прогноз життя пацієнта, відіграють злоякісні шлуночкові порушення ритму, найбільш характерні для АДПШ.
1. Аритмии сердца. Механизмы, диагностика, лечение / Под ред. В.Дж. Мандела. — М.: Медицина, 1996.
2. Кушаковский М.С., Журавлева Н.Б. Аритмии и блокады сердца (атлас электрокардиограмм). — Л.: Медицина, 1981.
3. Руководство по нарушениям ритма сердца / Под ред. Е.И. Чазова, С.П. Голицына. — М.: ГЭОТАР-Медиа, 2008.
4. ACC/AHA/ESC, 2006. Guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death // Circulation. — Vol. 114. — P. e385-e484.
5. Corrado D., Buja G., Basso C. et al. Clinical diagnosis and management strategies in arrhythmogenic right ventricu lar cardiomyopathy // J. Electrocardiol. — 2000. — № 33. — P. 49-55.
6. Fontaine G., Guiraudon G., Frank R. et al. Stimulation studies and epicardial mapping in ventricular tachycardia: Study of mechanisms and selection for surgery // Reentrant Arrhythmias / Ed. by H.E. Kulbertus. — Lancaster: MTP Press, 1977. — P. 334-350.
7. Theine G., Nava A., Corrado D. et al. Right ventricular cardiomyopathy and sudden death in young people // N. Engl. J. Med. — 1988. — № 318. — P. 129-133.
8. Report of the WHO/ISFC Task Force on the Definition and Classifica tion of Cardiomyopathy // Br. Heart J. — 1980. — № 44. — P. 672-673.
9. Rampazzo A., Nava A., Danieli G.A. et al. The gene for arrhythmogenic right ventricular cardiomiopathy maps to chromosome 14q23q24 // Hum. Mol. Genet. — 1994. — № 3. — P. 959-962.
10. Rampazzo A., Nava A., Erne P. et al. A locus for arrhythmogenic right ventricular cardiomiopathy (ARVD2) maps to chromosome Iq42q43 // Hum. Mol. Genet. — 1995. — № 4. — P. 2151-2154.
11. Tiso N., Stephan D.A., Nava A. et al. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ven tricular cardiomiopathy type 2 (ARVD2) // Hum. Mol. Genet. — 2001. — № 10. — P. 189-194.
12. Coonar A.S., Protonotarios N., Tsatsopoulou A. et al. Cardiac abnor malities in familial palmoplantar kera tosis // Br. Heart J. — 1986. — № 56. — P. 321-326.
13. Marcus F., Towbin J., Zareba W. et al. Arrhythmogenic right ventricular dysplasia/cardiomiopathy (ARVD/C). A multidisciplinary study: design and pro tocol // Circulation. — 2003. — № 107. — P. 2975-2978.
14. Fontaine G., Fontaliran F., Herbert J.L. et al. Arrhythmogenic right ventricular dysplasia // Ann. Rev. Med. — 1999. — № 50. — P. 17-35.
15. Marcus F., Fontaine G. Arrhythmogenic right ventricular dysplasia/cardiomyopathy: a review // Pacing Clin. Electrophysiol. — 1995. — № 18. — P. 1298-314.