
Газета «Новости медицины и фармации» 21 (349) 2010
Вернуться к номеру
Национальное соглашение: идиопатические интерстициальные пневмонии — классификация, диагностика, лечение*
Авторы: Ю.И. Фещенко, В.К. Гаврисюк, Н.Е. Моногарова, С.И. Лещенко, А.И. Ячник, И.В. Лискина, Национальный институт фтизиатрии и пульмонологии им. Ф.Г. Яновского НАМН Украины, Донецкий национальный медицинский университет им. М. Горького
Версия для печати
Разработка проекта основана на материалах международных соглашений American Thoracic Society, European Respiratory Society. Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement, 1999 [15] и American Thoracic Society/European Respiratory Society. International Multidisciplinary Consensus on the Idiopathic Interstitial Pneumonias, 2001 [16], а также многолетнем опыте обследования и лечения больных в Национальном институте фтизиатрии и пульмонологии имени Ф.Г. Яновского АМН Украины.
Определение
Идиопатические интерстициальные пневмонии (ИИП) — это группа заболеваний легких неустановленной этиологии, отличающихся друг от друга патоморфологическим типом неинфекционного воспаления и фиброза преимущественно в интерстиции легкого, а также вариантом клинического течения и прогноза — от острого с летальным исходом, хронического с формированием «сотового легкого» и нарастающей легочной недостаточностью до благоприятного, вплоть до клинического излечения.
ИИП являются одной из подгрупп среди диффузных паренхиматозных заболеваний легких (синоним — интерстициальные заболевания легких). Это гетерогенная группа неопухолевых поражений легких в результате диффузного повреждения легочной паренхимы с ее разнообразной структурной патологической перестройкой [16, 33].
Интерстиций представляет собой пространство между базальными мембранами эпителиального покрова альвеол и эндотелиальных клеток сосудов капиллярного русла, расположенных в межальвеолярных перегородках, и является первичной анатомической структурой-мишенью повреждения в патогенезе ИИП [16, 21, 29]. Однако повреждению часто подвергаются не только собственно интерстиций, но и альвеолярные пространства, структуры периферических воздухоносных путей, сосуды, расположенные вдоль вовлеченных в патологический процесс базальных мембран [16].
Термин «идиопатические» обозначает факт неизвестной причины возникновения патологии, а словосочетание «интерстициальная пневмония» характеризует преимущественное вовлечение в патологический процесс именно легочной паренхимы с различными количественными комбинациями фиброза и воспаления, в отличие от поражения преимущественно воздухосодержащих пространств, что характерно для банальной бактериальной пневмонии. В настоящее время синонимами являются также термины «идиопатический» и «криптогенный».
Эпидемиология ИИП
В литературе отсутствуют данные о распространенности отдельных форм ИИП. Исключение составляет идиопатический фиброзирующий альвеолит (ИФА) — наиболее частая форма ИИП (до 80 % всех случаев). По сведениям Американского торакального общества (ATS) [34], распространенность ИФА достигает 20,2 случая на 100 тыс. среди мужчин и 13,2 — среди женщин. Заболеваемость составляет в среднем 11,3 случая в год на 100 тыс. у мужчин и 7,1 — у женщин [19].
В последние годы проведены масштабные эпидемиологические исследования в США [35], Великобритании [22], Финляндии [25] и Норвегии [40]. Согласно полученным данным, распространенность ИФА в США составляет в среднем 14,0 на 100 тыс., а заболеваемость — 6,8 на 100 тыс. населения. В Великобритании заболеваемость ИФА составляет в среднем 4,6 на 100 тыс. Таким образом, ИФА не относится к категории редких заболеваний.
Показатели заболеваемости и распространенности ИФА значительно зависят от возраста. Так, если в возрастной группе от 18 до 34 лет заболеваемость ИФА составляет 0,4 на 100 тыс., то среди лиц в возрасте 75 лет и старше — 27,1 на 100 тыс.; распространенность — 0,8 и 64,7 на 100 тыс. соответственно [35]. За последние годы число больных ИФА увеличивается. Например, распространенность ИФА в Норвегии за семь лет возросла с 19,7 до 23,9 на 100 тыс. населения [40].
Классификация
В 2001 году было принято международное соглашение Американского торакального общества (ATS) и Европейского респираторного общества (ERS) [16], в котором приведена клинико-морфологическая характеристика 7 типов ИИП:
1) идиопатический легочный фиброз (в Украине и в большинстве стран постсоветского пространства в качестве синонима используется термин «идиопатический фиброзирующий альвеолит» (ИФА), в Европе, особенно в Великобритании, распространен термин «криптогенный фиброзирующий альвеолит»);
2) неспецифическая интерстициальная пневмония;
3) криптогенная организующая пневмония;
4) острая интерстициальная пневмония;
5) респираторный бронхиолит, ассоциированный с интерстициальным заболеванием легких;
6) десквамативная интерстициальная пневмония;
7) лимфоидная интерстициальная пневмония.
Современная классификация ИИП основана на учете особенностей клинической картины, рентгенологических и патоморфологических признаков. Исследование функции внешнего дыхания не позволяет выявить нарушения, патогномоничные для каждой формы ИИП. У всех пациентов наблюдаются рестриктивные нарушения легочной вентиляции — уменьшение общей емкости легких за счет ее составляющих. Исключение составляет респираторный бронхиолит, ассоциированный с интерстициальным заболеванием легких, при котором имеют место обструктивные нарушения с увеличением остаточного объема легких. В результате анализа газового состава и кислотно-основного состояния крови у больных определяются гипоксемия и гипокапния с дыхательным алкалозом, что характерно для большинства интерстициальных болезней легких.
Диагностика
Особенности клинических и рентгенологических проявлений различных форм ИИП
Идиопатический фиброзирующий альвеолит
Клинические симптомы. Идиопатический фиброзирующий альвеолит (ИФА) обычно проявляется постепенно нарастающей одышкой и непродуктивным кашлем, который нередко имеет приступообразный характер и отличается рефрактерностью к противокашлевым средствам. Среди больных преобладают мужчины в возрасте старше 50 лет. Деформация ногтевых фаланг в виде «барабанных палочек» отмечается у 25–50 % пациентов. При аускультации феномен «треск целлофана» в конце выдоха определяется в нижних отделах, а затем над всей поверхностью легких. В зарубежной литературе используется другой термин — Velcro-type crackles, что означает «треск открывающейся застежки-липучки».
Признаки хронического легочного сердца (периферические отеки) могут наблюдаться на поздних стадиях заболевания.
У большинства пациентов период от начала появления симптомов до обращения к врачу превышает 6 месяцев. Средняя продолжительность жизни со времени установления диагноза составляет от 2,5 до 3,5 года [36].
Клиническое течение ИФА характеризуется постепенным ухудшением состояния больных, однако нередко наступает резкое прогрессирование, связанное с вирусной инфекцией, развитием пневмонии или диффузного альвеолярного повреждения [7].
Рентгенологические признаки. При рентгенографии легких наиболее часто наблюдаются периферические ретикулярные тени преимущественно в базальных отделах, связанные с формированием сотовых изменений в легочной ткани и уменьшением объема нижних долей. Вместе с тем в среднем 16 % пациентов с гистологически доказанным ИФА могут иметь неизмененную рентгенологическую картину. Число диагностических ошибок при анализе рент- генограмм достигает 50 % [14].
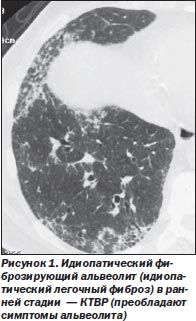 При компьютерной томографии высокого разрешения (КТВР) определяются ретикулярные изменения, обычно двухсторонние, отчасти связанные с тракционными бронхоэктазами. Часто наблюдаются признаки формирования «сотового легкого». Участки «матового стекла» распространены в меньшей степени, чем ретикулярные изменения [28, 31]. Характерны нарушения архитектоники, отражающие легочный фиброз. Патологические изменения характеризуются неоднородностью и локализованы преимущественно в периферических и базальных отделах [18] (рис. 1).
При компьютерной томографии высокого разрешения (КТВР) определяются ретикулярные изменения, обычно двухсторонние, отчасти связанные с тракционными бронхоэктазами. Часто наблюдаются признаки формирования «сотового легкого». Участки «матового стекла» распространены в меньшей степени, чем ретикулярные изменения [28, 31]. Характерны нарушения архитектоники, отражающие легочный фиброз. Патологические изменения характеризуются неоднородностью и локализованы преимущественно в периферических и базальных отделах [18] (рис. 1).
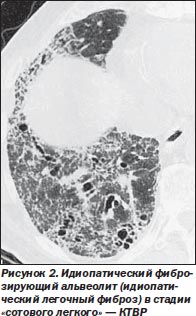
В ряде исследований, проведенных в процессе лечения больных, установлено, что зоны «матового стекла» могут уменьшаться. Однако наиболее характерно прогрессирование фиброза с формированием «сотового легкого» [13] (рис. 2). Точность диагностики ИФА по данным КТВР достигает 90 %.
 Неспецифическая интерстициальная пневмония
Неспецифическая интерстициальная пневмония
Клинические симптомы. По сравнению с ИФА неспецифическая интерстициальная пневмония (НСИП) развивается в более молодом возрасте (в среднем от 40 до 50 лет), одинаково часто у мужчин и женщин. Не связана с курением.
Заболевание начинается постепенно, у небольшой части больных возможно подострое начало. Средняя продолжительность существования симптомов до установления диагноза — от 1,5 до 3 лет [20, 39].
Клиническая картина НСИП сходна с таковой при ИФА, но одышка и кашель менее выражены и не нарастают столь неуклонно [3]. Примерно у половины больных отмечается уменьшение массы тела (в среднем до 6 кг). Повышение температуры тела наблюдается относительно редко, изменения ногтевых фаланг — в среднем у 10–35 % больных. При исследовании ФВД определяются незначительные или умеренно выраженные рестриктивные расстройства легочной вентиляции, снижение диффузионной способности легких, при нагрузке возникает гипоксемия.
В большинстве случаев НСИП хорошо поддается лечению глюкокортикостероидами (ГКС) и имеет благоприятный прогноз вплоть до клинического излечения.
Рентгенологические признаки.
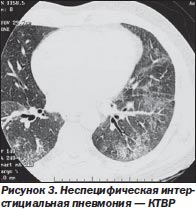 Рентгенография выявляет билатеральные инфильтративные изменения в нижних отделах легких. На КТВР наиболее часто определяются симметричные субплеврально расположенные участки «матового стекла» (рис. 3). У одной трети больных этот симптом является единственным проявлением заболевания. Ретикулярные изменения наблюдаются примерно в половине случаев. Признаки «сотового легкого», участки уплотнения легочной ткани отмечаются относительно редко. При повторных исследованиях в процессе лечения у большинства больных отмечается положительная рентгенологическая динамика.
Рентгенография выявляет билатеральные инфильтративные изменения в нижних отделах легких. На КТВР наиболее часто определяются симметричные субплеврально расположенные участки «матового стекла» (рис. 3). У одной трети больных этот симптом является единственным проявлением заболевания. Ретикулярные изменения наблюдаются примерно в половине случаев. Признаки «сотового легкого», участки уплотнения легочной ткани отмечаются относительно редко. При повторных исследованиях в процессе лечения у большинства больных отмечается положительная рентгенологическая динамика.
Криптогенная организующая пневмония
Криптогенная организующая пневмония (КОП) характеризуется вовлечением в патологический процесс дистальных воздушных пространств — альвеолярных ходов и альвеол в сочетании с полипоидным бронхиолитом или без него.
КОП одинаково часто встречается у мужчин и женщин. Средний возраст начала болезни — 55 лет. Симптомы заболевания обычно сохраняются менее 3 мес. Характерно гриппоподобное начало болезни: кашель, лихорадка, миалгии, недомогание [27]. Кашель может быть продуктивным, с выделением прозрачной бесцветной мокроты. В легких выслушиваются локализованные или распространенные трескучие хрипы. Форма ногтевых фаланг не изменяется.
Симптомы обычно расцениваются как проявление инфекции нижних дыхательных путей, в связи с чем многим больным безуспешно проводится антибиотикотерапия.
При лабораторном исследовании крови часто выявляется повышение содержания С-реактивного белка, нейтрофилов, увеличение СОЭ. При исследовании ФВД определяются умеренные рестриктивные расстройства, снижение диффузионной способности легких, возможна небольшая артериальная гипоксемия.
При назначении ГКС у большинства больных наступает полное выздоровление. Однако в период от 1 до 3 мес. после прекращения ГКС-терапии или уменьшения дозы ниже 15 мг в сутки часто наблюдаются рецидивы заболевания. В связи с этим продолжительность ГКС-терапии должна быть не менее 6 мес.
Рентгенологические признаки.
Наиболее характерными рентгенологическими проявлениями заболевания являются билатеральные или односторонние латеральные затемнения [30]. Небольшие узелковые тени отмечаются в 10–50 % случаев, более крупные узелковые образования ( > 1 см) наблюдаются приблизительно у 15 % больных. Ретикуло-узелковый паттерн рентгенологических изменений регистрируется относительно редко. Уменьшение площади легочных полей отмечается в 25 % случаев.
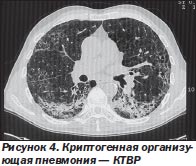 На КТВР в 90 % случаев определяются субплевральные и перибронхиальные уплотнения легочной ткани, чаще в нижних долях легких (рис. 4). Небольшие узелки, расположенные вдоль бронхососудистых пучков, наблюдаются менее чем в 50 % случаев. У 60 % больных имеются участки «матового стекла».
На КТВР в 90 % случаев определяются субплевральные и перибронхиальные уплотнения легочной ткани, чаще в нижних долях легких (рис. 4). Небольшие узелки, расположенные вдоль бронхососудистых пучков, наблюдаются менее чем в 50 % случаев. У 60 % больных имеются участки «матового стекла».
У большинства больных в процессе лечения отмечается улучшение рентгенологической картины. У пациентов, которые не лечились ГКС, паренхимальные изменения могут регрессировать в одних зонах легких, одновременно возникая в других участках.
Острая интерстициальная пневмония
Острая интерстициальная пневмония (ОИП) (синдром Хаммена — Рича) — редкая быстропрогрессирующая форма диффузного альвеолярного организующегося повреждения легких. Патоморфологически это одна из форм диффузного альвеолярного повреждения (ДАП), которая не имеет отличий от гистологического паттерна острого респираторного дистресс-синдрома при сепсисе или шоке.
Клинические симптомы. Заболевание может развиться в любом возрасте и с одинаковой частотой у мужчин и женщин, вне зависимости от курения. Развитию тяжелой нарастающей одышки часто предшествуют симптомы — миалгии, артралгии, лихорадка, озноб, недомогание [26]. В легких выслушиваются распространенные «целлофановые» хрипы. Через несколько дней развивается тяжелая одышка, цианоз.
Пульмональные функциональные тесты демонстрируют рестриктивный тип нарушений вентиляции в сочетании с расстройствами диффузионной способности легких. Легочная недостаточность быстро прогрессирует, при этом часто наблюдается рефрактерность к оксигенотерапии [32]. Как правило, больным требуется искусственная вентиляция легких.
Дифференциальная диагностика должна проводиться между ОИП и ДАП при остром респираторном дистресс-синдроме, при коллагеновых болезнях, инфекциях (особенно пневмонии, вызванной Pneumocystis carinii и цитомегаловирусом), лекарственно обусловленных пневмонитах, острой эозинофильной пневмонии, гиперсенситивном пневмоните.
Лечение ГКС и цитостатиками малоэффективно. Смертность превышает 50 %, большинство больных умирает в течение 1–2 мес. после появления симптомов [26]. У выживших пациентов могут быть рецидивы или развитие прогрессирующего интерстициального заболевания легких.
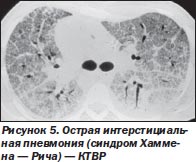 Рентгенологические признаки. На рентгенограмме выявляются диффузные билатеральные затемнения, которые имеют неоднородный пятнистый характер. Плевральный выпот обычно отсутствует. При КТВР видны двухсторонние симметричные, преимущественно субплеврально расположенные негомогенные области «матового стекла», участки уплотнения воздушных пространств, расширение бронхиол на фоне нарушения нормальной архитектоники легких (рис. 5).
Рентгенологические признаки. На рентгенограмме выявляются диффузные билатеральные затемнения, которые имеют неоднородный пятнистый характер. Плевральный выпот обычно отсутствует. При КТВР видны двухсторонние симметричные, преимущественно субплеврально расположенные негомогенные области «матового стекла», участки уплотнения воздушных пространств, расширение бронхиол на фоне нарушения нормальной архитектоники легких (рис. 5).
Позднее, на организующей стадии ОИП, появляются нарушения структуры бронхососудистых пучков, тракционные бронхоэктазы. У пациентов, которые перенесли острейшую фазу болезни, наблюдается постепенное уменьшение участков уплотнения и «матового стекла», при этом могут формироваться изменения сетчатого характера.
Респираторный бронхиолит, ассоциированный с интерстициальным заболеванием легких
Респираторный бронхиолит, ассоциированный с интерстициальным заболеванием легких (РБ-ИЗЛ) характеризуется поражением респираторных бронхиол с наличием пигментированных макрофагов в их стенках, сочетающимся с интерстициальным заболеванием легких.
Клинические симптомы. РБ-ИЗЛ — болезнь курильщиков со стажем более 30 лет. У большинства пациентов симптомы заболевания выражены незначительно, но у части больных могут развиваться тяжелая одышка и гипоксемия. Заболевание начинается постепенно: появляется или усиливается кашель, начинает беспокоить одышка. При физикальном обследовании патологические изменения в легких часто не определяются, у части больных могут выслушиваться трескучие хрипы.
Отличительной чертой этой формы ИИП при исследовании ФВД является наличие не только рестриктивных, но и обструктивных нарушений легочной вентиляции с увеличением остаточного объема легких. Отмечается также умеренное снижение диффузионной способности легких.
Клиническое течение и прогноз РБ-ИЗЛ чаще благоприятны. Прекращение курения, как правило, обусловливает уменьшение выраженности одышки. Терапия ГКС эффективна.
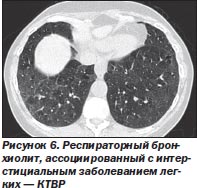 Рентгенологические признаки. Наиболее характерные изменения на рентгенограмме — утолщение стенок центральных и периферических бронхов (75 % больных), участки матового стекла (60 %). Примерно у 14 % больных рентгенограмма соответствует норме [24].
Рентгенологические признаки. Наиболее характерные изменения на рентгенограмме — утолщение стенок центральных и периферических бронхов (75 % больных), участки матового стекла (60 %). Примерно у 14 % больных рентгенограмма соответствует норме [24].
При КТВР определяются центрилобулярные узелки, распространенные участки «матового стекла», утолщение стенок бронхов, признаки центрилобулярной эмфиземы в легких (рис. 6).
Десквамативная интерстициальная пневмония
Десквамативная интерстициальная пневмония (ДИП) является близкой к РБ-ИЗЛ формой идиопатических пневмоний по характеру патоморфологических изменений в легких и клиническим проявлениям. Отличается накоплением в альвеолах макрофагов.
Клинические симптомы. ДИП встречается редко (< 3 % всех случаев ИИП), преимущественно у курящих мужчин 40–50 лет. У большинства пациентов заболевание протекает подостро в течение нескольких недель или месяцев, проявляется сухим кашлем и нарастающей одышкой. При исследовании ФВД выявляются умеренные рестриктивные нарушения, снижение диффузионной способности легких. ГКС-терапия достаточно эффективна, прогноз благоприятен.
Рентгенологические признаки. На рентгенограмме преобладает симптом «матового стекла» преимущественно в нижних отделах легких. Описана также узелковая текстура участков «матового стекла».
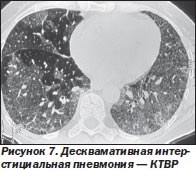 При КТВР участки «матового стекла» определяются во всех случаях [23]. В нижних зонах довольно часто определяются линейные и ретикулярные тени, возможно формирование ограниченных субплевральных участков «сотового легкого» (рис. 7).
При КТВР участки «матового стекла» определяются во всех случаях [23]. В нижних зонах довольно часто определяются линейные и ретикулярные тени, возможно формирование ограниченных субплевральных участков «сотового легкого» (рис. 7).
Лимфоидная интерстициальная пневмония
Клинические симптомы. Лимфоидная интерстициальная пневмония (ЛИП) встречается редко, обычно у женщин, чаще после 40 лет. Заболевание развивается медленно, одышка и кашель постепенно нарастают в течение 3 лет и более. Характерны лихорадка, боль в груди, артралгии, похудание. В легких выслушиваются трескучие хрипы. Могут наблюдаться анемия, гипергаммаглобулинемия.
Заболевание поддается терапии ГКС и имеет благоприятный прогноз, однако примерно у 1/3 пациентов формируется диффузный интерстициальный фиброз.
Рентгенологические признаки. При рентгенографии легких могут наблюдаться два типа изменений: нижнедолевые смешанные альвеолярно-интерстициальные инфильтраты и диффузное поражение с формированием «сотового легкого».
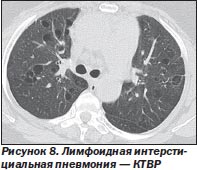 На КТВР обычно определяются участки «матового стекла». Иногда определяются периваскулярные кисты и участки «сотового легкого» (рис. 8). Изменения ретикулярного характера наблюдаются примерно в 50 % случаев.
На КТВР обычно определяются участки «матового стекла». Иногда определяются периваскулярные кисты и участки «сотового легкого» (рис. 8). Изменения ретикулярного характера наблюдаются примерно в 50 % случаев.
Роль хирургической биопсии легких и патоморфологическая диагностика ИИП
Хирургическая биопсия легких, открытая или видеоторакоскопическая, необходима для установления достоверного клинико-патоморфологического диагноза, за исключением случаев типичной клинико-рентгенологической картины ИФА.
Целесообразность проведения хирургической биопсии легких определяется следующим.
1. Установление достоверного клинико-патоморфологического диагноза позволяет принять более информированное решение относительно лечения больного.
2. Терапия ИИП имеет потенциально серьезные риски развития побочных явлений, и подвергать пациентов этим рискам в случае неуверенности в диагнозе неприемлемо.
3. Определение в результате биопсии фиброзного процесса в легких, имеющего отношение к воздействию специфических факторов (например, асбестоз), может иметь важное компенсационное значение для пациента.
Биопсия позволяет подтвердить или исключить альтернативные диагнозы, такие как саркоидоз, гиперсенситивный пневмонит, эозинофильная пневмония, альвеолярная карцинома, лимфома, гистиоцитоз Х.
Для морфологической верификации ИИП требуется получение достаточно больших образцов легочной ткани, что невозможно при трансбронхиальной биопсии. Вместе с тем бронхоскопическая биопсия позволяет во многих случаях подтвердить или исключить саркоидоз, неоплазму, инфекционный процесс, что имеет значение в диагностике ИИП.
Каждая форма ИИП имеет свой гистологический паттерн [3]. Основные патоморфологические различия идиопатических пневмоний представлены в табл. 1.
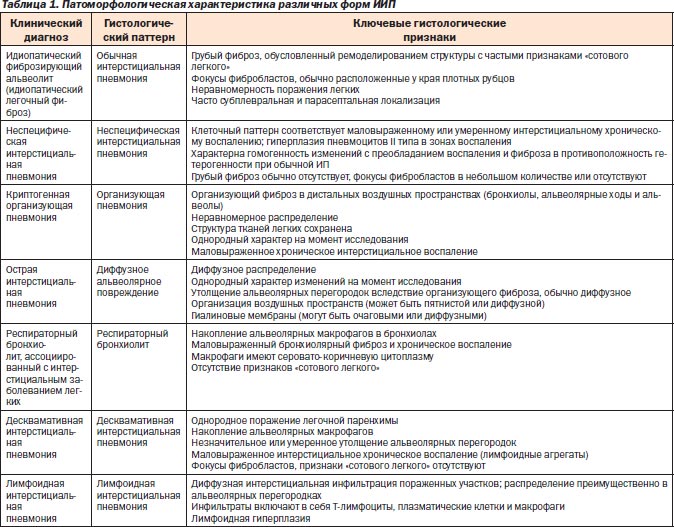
Морфологическая идентификация идиопатических интерстициальных пневмоний трудна, в связи с чем специалист-патоморфолог в своей работе должен учитывать и клинические данные. Выбор оптимальных размеров биоптатов и числа долей легких, подлежащих биопсии, проводится при участии пульмонолога, рентгенолога, патоморфолога и хирурга. На этом этапе диагностики возникают деонтологические вопросы, касающиеся оправданности применения инвазивного метода исследования. Для их решения всегда надо сравнивать ущерб, наносимый больному методом исследования, и возможные последствия неточной диагностики и ошибок в лечении.
Показания к использованию биопсии легкого:
— невозможность установления диагноза без ее применения;
— необходимость выбора терапии;
— отсутствие признаков «сотового легкого» — конечной фазы многих интерстициальных болезней легких.
Для случаев ИФА, когда хирургическая биопсия легкого рискованна, разработаны критерии диагностики, позволяющие с высокой вероятностью установить диагноз, не прибегая к гистологической верификации (табл. 2).
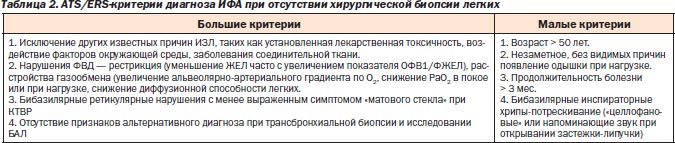
Лечение
Фундаментальные подходы к лечению больных ИФА, основанные на принципах агрессивной противовоспалительной терапии, остаются неизменными на протяжении последних 30 лет.
Глюкокортикостероиды (ГКС) и цитостатики — основные компоненты лечения больных ИФА, несмотря на то что у значительной части пациентов эти препараты не оказывают существенного влияния на продолжительность жизни. До настоящего времени еще нет средства, способного остановить воспалительный процесс или процессы фиброзирования при ИФА.
Принято считать, что у 10–40 % больных ИФА начальная терапия с использованием ГКС приводит к частичному улучшению состояния, при этом полная ремиссия заболевания наблюдается в единичных случаях. Вместе с тем результаты сравнительного наблюдения больных на фоне лечения и больных, по ряду причин не принимавших ГКС, свидетельствуют, что у последних ни в одном случае не отмечалось улучшения в течении ИФА. Таким образом, при этом заболевании не наблюдаются спонтанные ремиссии, а средние сроки жизни, по последним данным, составляют от 2 до 4 лет от момента установления диагноза. В связи с этим, несмотря на неблагоприятный прогноз для большей части больных ИФА, лечение должно быть назначено всем пациентам, у которых нет противопоказаний к ГКС или цитостатикам.
Вместе с тем необходимо учитывать, что ожидаемый лечебный эффект должен перевешивать возрастающий риск осложнений от терапии. Прежде всего это относится к пациентам в возрасте старше 70 лет, больным с крайней степенью ожирения, сопутствующими тяжелыми заболеваниями сердца и сосудов, сахарным диабетом, остеопорозом, тяжелыми нарушениями функции внешнего дыхания, признаками сформировавшегося «сотового легкого» при рентгенологическом исследовании.
Большинство специалистов рекомендуют высокие дозы ГКС: перорально 1 мг преднизолона на 1 кг так называемой тощей массы тела (без учета подкожной жировой клетчатки — в среднем на 15 % меньше фактической), но не более 60 мг в сутки. Эту дозу назначают в течение 2–4 мес. с последующим снижением до поддерживающей — 15–20 мг/сут. Цитостатическая терапия (циклофосфамид и азатиоприн) применялась ранее у больных ИФА, у которых отсутствовал ответ на ГКС-терапию, у пациентов с осложнениями или высоким риском осложнений от ГКС. В настоящее время считают, что комбинированное лечение ГКС и цитостатиками повышает эффективность и одновременно позволяет существенно снизить суммарные дозы тех и других препаратов.
Обычно рекомендуют применение 15–25 мг преднизолона ежедневно и 200 мг циклофосфана 2 раза в неделю.
Рекомендации ERS и ATS по лечению больных ИФА [15]:
— кортикостероид (преднизолон или аналог в эквивалентной дозе) — 0,5 мг/кг массы тела в день перорально в течение 4 недель; 0,25 мг/кг в день в течение 8 недель. Постепенное снижение до 0,125 мг/кг в день или 0,25 мг/кг через день;
— плюс азатиоприн — 2–3 мг/кг в день; максимальная доза — 150 мг в день. Лечение начинают с 25–50 мг в день, увеличивая дозу на 25 мг каждые 1–2 недели до достижения максимальной дозы;
— или циклофосфамид — 2 мг/кг в день. Максимальная доза — 150 мг в день. Лечение начинают с 25–50 мг в день, увеличивая дозу на 25 мг каждые 1–2 недели до достижения максимальной дозы.
Терапия должна продолжаться как минимум 6 месяцев. Эффективность определяется на основании оценки симптомов, рентгенологических и физиологических данных. Необходим тщательный мониторинг побочных эффектов терапии.
Начало лечения должно осуществляться на ранних стадиях заболевания, когда отсутствуют выраженные необратимые фиброзные изменения в паренхиме легких. Недостаточный эффект терапии часто обусловлен задержкой в назначении ГКС и цитостатиков.
Продолжительность лечения. Эффективность терапии может быть достаточно объективно оценена, если пациент принимает препараты на протяжении трех и более месяцев. Таким образом, при отсутствии осложнений или побочных явлений комбинированная терапия должна продолжаться по крайней мере 6 месяцев. В течение этого периода ежемесячно проводится клиническое наблюдение с оценкой динамики функциональных и рентгенологических данных.
Экспертами ERS и ATS разработаны следующие критерии оценки эффективности терапии больных ИФА [15]:
1. Клиническое улучшение: наличие не менее двух следующих критериев в течение двух последовательных визитов в период от 3 до 6 месяцев лечения:
— симптомы: уменьшение степени одышки и тяжести кашля;
— радиология: уменьшение паренхиматозных изменений по данным рентгенографии или КТВР легких;
— физиология: улучшение, определяемое наличием не менее двух следующих критериев:
– ≥ 10 % увеличение TLC или FVC (минимум 200 мл);
– ≥ 15 % увеличение DLco (минимум 3 мл/мин/мм Hg);
– значительное улучшение (≥ 4%- единиц, ≥ 4 мм Hg) SaO2 или РаО2, измеренных при проведении теста с физической нагрузкой.
2. Клиническая стабилизация: наличие не менее двух следующих критериев в течение двух последовательных визитов в период от 3 до 6 месяцев лечения:
— симптомы: нет значительных изменений;
— радиология: нет значительных изменений;
— физиология: стабилизация, определяемая наличием не менее двух следующих критериев:
– < 10 % изменение TLC или FVC;
– < 15 % увеличение DLco;
– нет значительных изменений SaO2 или РаО2, измеренных при проведении теста с физической нагрузкой.
3. Отсутствие эффекта (после 6 месяцев терапии):
— симптомы: увеличение степени одышки и тяжести кашля, не связанное с другими факторами;
— радиология: увеличение паренхиматозных изменений, или признаков «сотового легкого», или признаков легочной гипертензии, по данным рентгенографии или КТВР легких;
— физиология: ухудшение, определяемое наличием не менее двух следующих критериев:
– ≥ 10 % уменьшение TLC или FVC;
– ≥ 15 % уменьшение DLco;
– значительное ухудшение (≥ 4 %- единиц, ≥ 4 мм Hg) SaO2 или РаО2, измеренное при проведении теста с физической нагрузкой.
Примечания: КТВР — компьютерная томография высокого разрешения; DLco — диффузионная способность легких по оксиду углерода; FVC — форсированная жизненная емкость легких; РаО2 — напряжение артериальной крови кислородом; SaO2 — насыщение артериальной крови кислородом; TLC — общая емкость легких.
Важнейшим компонентом лечебного процесса является мониторинг нежелательных эффектов терапии.
До начала терапии пациенты должны быть информированы о потенциальном риске и побочных эффектах кортикостероидной и цитостатической терапии. Большинство больных переносят терапию ГКС удовлетворительно. Однако у части пациентов развиваются побочные эффекты и осложнения — пептические язвы желудка, катаракта, повышение внутриглазного давления, гипертензия, эндокринные и метаболические нарушения (ожирение, лунообразность лица, расстройства менструального цикла, импотенция, гипергликемия, гипокалиемия, метаболический алкалоз, вторичная недостаточность надпочечников). Серьезными осложнениями являются повреждения костно-мышечной системы — остеопороз, компрессионные переломы позвоночника, асептический некроз головки бедренной и плечевой кости, миопатия. В более поздние сроки лечения могут повреждаться диафрагмальная и межреберные мышцы, что осложняет оценку эффективности терапии. Психологические эффекты включают эйфорию, депрессию, у пожилых людей может развиться психоз. Особое значение имеют методы профилактики остеопороза у женщин в период менопаузы, у которых даже относительно короткий курс терапии ГКС (от 3 до 6 мес.) может вызвать уменьшение костной массы.
Лечение циклофосфамидом и азатиоприном требует еженедельного контроля количества в крови лейкоцитов и тромбоцитов. Если количество лейкоцитов уменьшается до уровня ≤ 4000/мм3, а содержание тромбоцитов падает ниже 100 000/мм3, лечение следует приостановить или немедленно уменьшить дозу на 50 %. Контроль темпов восстановления числа лейкоцитов и тромбоцитов проводится еженедельно. Если восстановления не наблюдается, цитостатики следует отменить до достижения нормализации клеточного состава крови.
Азатиоприн характеризуется более выраженным гепатотоксическим действием. В связи с этим пациентам, принимающим азатиоприн, необходимо ежемесячно осуществлять определение уровня трансаминаз. Лечение следует приостановить или уменьшить дозу в случаях, когда содержание аланинаминотрансферазы более чем в 3 раза превышает нормальный уровень.
У больных, принимающих циклофосфамид, иногда развивается геморрагический цистит. С целью его профилактики рекомендуется обильное питье с ежемесячным контролем количества эритроцитов в моче.
Кортикостероидная терапия может вызвать супрессию иммунного ответа при проведении кожных проб. В связи с этим кожную пробу с туберкулином, если она необходима, следует проводить до начала лечения ГКС. Ряд специалистов рекомендует использование у больных, принимающих иммуносупрессивную терапию, триметопримсульфаметаксазола как профилактического средства против Pneumocystis carinii (по одной большой таблетке 3 раза в неделю) и изониазида — против Mycobacterium tuberculosis. Применение изониазида рекомендуется лицам, относящимся к группам риска по туберкулезу.
Таким образом, терапия ГКС и цитостатиками сопряжена с риском развития серьезных побочных явлений и осложнений. Однако следует помнить, что спонтанных ремиссий при ИФА практически не бывает, а альтернативы ГКС и цитостатическим средствам в настоящее время не существует. В связи с этим и лечащий врач, и больной должны быть готовы к возможным нежелательным эффектам терапии. При этом необходимо учесть, что нет универсальных схем борьбы с нежелательными эффектами кортикостероидной и цитостатической терапии. В каждом конкретном случае необходим индивидуальный подход, и только искусство врача, понимание и терпение больного могут быть залогом успеха.
Наличие побочных эффектов циклофосфамида и азатиоприна явилось предпосылкой для изучения эффективности в лечении больных ИФА других цитостатических средств — циклоспорина А, метотрексата, хлорамбуцила.
Опыт применения циклоспорина А при ИФА весьма ограничен. Литература не содержит описания случаев достижения устойчивой ремиссии заболевания, при этом отмечена высокая токсичность препарата. Метотрексат успешно применяется при ряде иммунозависимых поражений легких. Вместе с тем известна потенциальная легочная токсичность препарата при длительном использовании, что уменьшает перспективы его применения при ИФА. Хлорамбуцил характеризуется токсическим поражением желудка, кишечника и костного мозга, может индуцировать новообразования (в том числе лейкемию). По этой причине препарат не рекомендуется для лечения больных ИФА.
Эффективность препаратов, тормозящих синтез коллагена и фиброзирование (D-пеницилламин, колхицин, интерфероны), до настоящего времени не доказана. Следует отметить, что D-пеницилламин (купренил), широко применявшийся ранее, более чем у половины больных вызывает серьезные побочные эффекты, которые по тяжести течения конкурируют с основным заболеванием.
В 2005 году были обобщены результаты рандомизированного двойного слепого плацебо-контролируемого исследования IFIGENIA, продемонстрировавшего целесообразность присоединения к стандартной терапии ГКС у больных ИФА N-ацетилцистеина в высоких дозах (1800 мг в сутки) [4, 8]. В этом исследовании, лидирующем в последние годы по количеству включенных больных с подтвержденным диагнозом ИФА (n = 155), больным в дополнение к ГКС и азатиоприну назначали N-ацетилцистеин или плацебо. Через 12 мес. лечения стало очевидным, что N-ацетилцистеин замедляет снижение жизненной емкости легких и диффузионной способности легких. Препарат позволяет более успешно, чем только иммуносупрессивная терапия, стабилизировать клинико-функциональные показатели у больных ИФА и смягчить нежелательные эффекты, вызываемые ГКС и цитостатиками.
В комплексе с ГКС и цитостатиками рекомендуется применение препаратов, содержащих омега-3 полиненасыщенные жирные кислоты (ПНЖК), которые в настоящее время применяются в комплексном лечении больных системными заболеваниями соединительной ткани, хроническим гломерулонефритом, псориатическим артритом. В механизме противовоспалительного действия омега-3 ПНЖК основное значение имеет влияние на метаболизм арахидоновой кислоты с уменьшением продукции лейкотриенов 4-й серии — активных провоспалительных факторов. Суточная лечебная доза омега-3 ПНЖК составляет 0,8–1,0 г (2,0 г препарата, содержащего 45 % омега-3 ПНЖК), длительность лечения соответствует продолжительности основного курса терапии.
При других формах идиопатических интерстициальных пневмоний, за исключением острой, в большинстве случаев достаточно применения монотерапии ГКС в начальной дозе 0,5 мг/кг. О наличии или отсутствии положительного эффекта ГКС можно судить уже в течение первых 1–2 недель лечения, ориентируясь на динамику клинических данных. При неэффективности ГКС к терапии необходимо добавить цитостатик, то есть использовать схему лечения больных ИФА. Решение о применении комбинированной терапии должно быть обоснованным, вместе с тем не следует допускать промедления в назначении активной терапии, поскольку каждый день неадекватного лечения уменьшает шансы больного на выздоровление.
Острая интерстициальная пневмония (синдром Хаммана — Рича) встречается весьма редко. Больные, в связи с крайне тяжелым состоянием, госпитализируются, как правило, в отделение интенсивной терапии и реанимации. В лечении больных острой интерстициальной пневмонией необходимо применять высокие дозы ГКС — внутривенную пульс-терапию (метилпреднизолон 1–2 г в сутки) в комбинации с методами лечения синдрома острой дыхательной недостаточности.
В поздних стадиях ИФА, вследствие редукции сосудистого русла и расстройств вентиляционно-перфузионных отношений, у большинства больных развивается гиперкапния, компенсаторный эритроцитоз, что обусловливает расстройства реологических свойств крови, в результате чего еще в большей мере нарушается газообмен в легких, повышается легочно-артериальное сопротивление. На этой стадии болезни рекомендуется применение инфузионных препаратов на основе раствора сорбитола (по 200 мл внутривенно капельно), обладающих гемодилюционным свойством. За счет более высокой осмолярности эти препараты обеспечивают активную дегидратацию тканей, что имеет важное значение при наличии воспалительного отека стенок альвеол. В результате уменьшается гематокрит, улучшается микроциркуляция в легких.
У больных с высоким эритроцитозом (гемоглобин — 170–180 г/л и более) введение инфузионных растворов рекомендуется сочетать с дозированным отбором крови (по 50 мл на одно введение при общем количестве инфузий 7–8).
Важнейшим методом терапии больных ИФА на стадии сформированного «сотового легкого» является длительная кислородотерапия.
Показания к длительной кислородотерапии:
— РаО2 < 55 мм рт.ст. или SaO2 < 88 % в покое;
— РаО2 < 56 — 59 мм рт.ст. или SaO2 — 89 % при наличии легочного сердца или эритроцитоза (Ht > 55 %).
Задачей кислородотерапии является коррекция гипоксемии и достижение значений РаО2 > 60 мм рт.ст. и SaO2 > > 90 %. Оптимальным считается поддержание РаО2 в пределах 60–65 мм рт.ст. и SaO2 в пределах 90–95 %. Обязательным условием кислородотерапии является мониторинг состояния оксигенации при помощи пульсоксиметрии или газового анализа артериальной крови.
Величина потока О2 составляет от 1 до 5 л/мин в зависимости от степени оксигенации крови. Рекомендуется проведение кислородотерапии не менее 15 часов в сутки. Максимальные перерывы между сеансами не должны превышать 2 ч.
В ночное время, при физической нагрузке необходимо увеличить поток О2 в среднем на 1 л/мин по сравнению с оптимальным дневным потоком. Для проведения длительной кислородотерапии в домашних условиях требуются автономные и портативные источники О2: концентраторы, баллоны со сжатым газом и резервуары с жидким О2.
В качестве систем для доставки О2 в домашних условиях чаще всего используются носовые канюли. Доставка О2 в альвеолы происходит только во время ранней фазы вдоха (примерно 1/6 часть дыхательного цикла), в то время как остальной О2 расходуется вхолостую. Для осуществления более эффективной доставки О2 существует несколько типов кислородосберегающих устройств: резервуарные канюли, пульсирующие устройства доставки О2 и транстрахеальные катетеры. При их использовании достигается экономия О2 в 2–4 раза.
Длительная кислородотерапия является дорогостоящим методом, вместе с тем в стадии «сотового легкого» это единственный способ продлить жизнь больного.
Практические рекомендации
Принятое ERS и ATS соглашение не может полностью разрешить все противоречия во взглядах на проблему ИИП. По мнению М.М. Ильковича и соавт. [2], все указанные выше идиопатические интерстициальные пневмонии следует рассматривать как единую нозологическую форму (ИФА), клинические проявления которой и течение (острое, подострое и хроническое) зависят от выраженности экссудации и пролиферации в тканях легких.
Основной причиной развития РБ-ИЗЛ и ДИП считают влияние табачного дыма, в связи с этим относить эти пневмонии к группе идиопатических, с нашей точки зрения, не вполне обоснованно.
Вместе с тем соглашение — это попытка хоть как-то упорядочить существовавшие разночтения. В настоящее время во всех областях знаний, в том числе и в медицине, наблюдается интенсивное развитие интеграционных процессов. Для проведения эпидемиологических исследований, многоцентровых клинических испытаний новых лекарственных препаратов необходимы единые подходы к пониманию сущности болезни, единые классификации, схемы диагностики и лечения. С этих позиций, несмотря на дискуссионный характер ряда положений, международное соглашение по ИИП, на наш взгляд, может составить основу для объединения усилий ученых и практических врачей разных стран в решении этой сложной проблемы в пульмонологии.
Несомненно, дифференцировать формы ИИП в практической работе пульмонолога весьма сложно. В связи с этим при подозрении на ИИП целесообразно придерживаться следующих рекомендаций.
Идентификация каждой формы ИИП не является самоцелью, она абсолютно необходима в случае, если точность диагностики определяет тактику лечения.
Применения максимальных доз ГКС требуют только две формы идиопатических пневмоний — ОИП (пульс-терапия до 1000 мг и более на одно введение в комбинации с полной дозой ГКС per os — 1 мг на 1 кг массы тела) и ИФА (полная доза при монотерапии и половинная — в комбинации с цитостатиками). Все остальные формы ИИП, а они составляют не более 15–20 % всех случаев ИИП, требуют применения ГКС, как правило, в средних дозах — 0,5 мг на 1 кг массы тела в расчете на преднизолон.
Установить с достаточно высокой степенью достоверности диагноз ИФА на основании клинических данных и результатов КТВР в большинстве случаев не так уж сложно, так как больные, как правило, обращаются к врачу в стадиях развернутой картины заболевания. Сегодня основной проблемой является ранняя диагностика ИФА. Решение именно этой проблемы позволит существенно увеличить продолжительность жизни больных.
ОИП встречается весьма редко и по клиническим проявлениям существенно отличается от других форм ИИП.
Идентификация остальных пяти форм ИИП представляет трудности и без применения хирургической биопсии, как правило, невозможна.
Таким образом, в случае ИФА пульмонолог, обладающий достаточным объемом знаний об ИИП и располагающий данными КТВР, с нашей точки зрения, должен установить клинический диагноз и безотлагательно назначить адекватную терапию. К сожалению, больные очень часто (на всякий случай) направляются на консультацию в противотуберкулезный диспансер, где назначается (на всякий случай) противотуберкулезная терапия на несколько месяцев, что несомненно усугубляет течение ИФА и укорачивает жизнь больных. Для уточнения диагноза больных необходимо направлять в Институт фтизиатрии и пульмонологии или в областные пульмонологические центры.
Диагноз ИФА складывается из названия болезни и указания степени легочной недостаточности (ЛН). Например: идиопатический фиброзирующий альвеолит, ЛН II степени. Если при КТВР выявляются признаки «сотового легкого», их необходимо указать в диагнозе в одном из двух вариантов — стадия формирования «сотового легкого» или стадия сформированного «сотового легкого» [1]. В случаях застойной недостаточности кровообращения (НК), дилатации правого желудочка сердца в диагнозе указывается наличие хронического легочного сердца и стадия НК.
После установления диагноза больному назначается лечение ГКС в высоких дозах или в средних — при сочетании с циклофосфамидом [15].
В соответствии с рекомендациями ATS/ERS [15], достоверная оценка эффективности лечения может быть проведена не ранее чем через 3 мес. от начала терапии, окончательная — через 6 мес. Это не означает, что следующий визит больного будет через 3 мес. после начала терапии. Весь период лечения должен проводиться постоянный мониторинг возможных побочных действий ГКС-терапии и лечения цитостатиками. Обследование больного (клиническое и спирометрию) необходимо проводить не реже 1 раза в 4 недели, а телефонный контакт — 1 раз в 2 недели.
Через 3 мес. после начала лечения и далее в диагнозе необходимо указывать результат проводимой терапии — фазу клинического улучшения, фазу стабилизации, фазу прогрессирования [15, 17]. Подчеркиваем, что эти формулировки имеют отношение только к оценке эффективности лечения, поскольку ИФА — заболевание изначально прогрессирующее.
Что касается синдрома Хаммена — Рича (ОИП), то пациенты с этой редкой формой ИИП обычно наблюдаются в практике реаниматолога, поскольку вследствие тяжести состояния они госпитализируются в отделение интенсивной терапии и реанимации и лечатся с диагнозом острого респираторного дистресс-синдрома. Исключение возможных причин ОРДС позволяет уверенно заподозрить идиопатический характер заболевания и назначить ГКС-терапию в адекватных дозах.
Идентификация НСИП, КОП, РБ-ИЗЛ, ДИП и ЛИП трудна, однако это не имеет существенного практического значения, поскольку все эти формы требуют единого подхода к лечению [5]. Если врач уверен, что обратившийся к нему пациент относится к группе больных ИИП и при этом нет оснований для диагноза ИФА, то диагноз одной из пяти вышеперечисленных форм пневмоний может быть установлен в предположительной форме, например: идиопатическая интерстициальная пневмония (неспецифическая), ЛН II степени. Больному необходимо назначить ГКС-терапию и направить его для уточнения диагноза в Институт фтизиатрии и пульмонологии или в областной пульмонологический центр.
В случаях, когда нет полной уверенности в отсутствии туберкулезного процесса, необходимо учитывать следующее. Пробная ГКС-терапия больных туберкулезом в течение короткого отрезка времени (10–12 дней) менее опасна, чем пробная терапия больных ИИП противотуберкулезными препаратами в течение нескольких недель, а то и месяцев. Быстрая положительная реакция на ГКС-терапию, что характерно для ИИП (за исключением ИФА), может снять проблему с диагнозом. С другой стороны, больной, госпитализированный в противотуберкулезный диспансер, автоматически попадает в группу риска по туберкулезу. В связи с этим, если у этого пациента впоследствии будет установлен диагноз ИИП, лечение ГКС необходимо сочетать с противотуберкулезным препаратом во избежание стероидного туберкулеза.
В заключение следует отметить, что эффективность лечения больных ИФА прежде всего зависит от сроков начала терапии: назначение противовоспалительных и цитостатических средств на ранней стадии заболевания существенно повышает эффективность лечения и улучшает прогноз. В связи с этим наиболее важной задачей в настоящее время является ранняя диагностика ИФА, что, в свою очередь, обусловливает необходимость повышения уровня знаний пульмонологов, терапевтов и рентгенологов об этом тяжелом заболевании.
1. Виноградова Д.Н., Амосов В.И., Илькович М.М. Идиопатический фиброзирующий альвеолит: возможности компьютерной томографии в первичном распознавании и уточнении стадии патологического процесса // Пульмонология. — 2003. — № 3. — С. 54-58.
2. Илькович М.М., Новикова Л.Н., Королева М.Г. Идиопатический фиброзирующий альвеолит: противоречия в современных представлениях // Пульмонология. — 2003. — № 3. — С. 98-101.
3. Лискина И.В., Моногарова Н.Е. Гистоморфологическая характеристика идиопатических интерстициальных пневмоний // Укр. пульмонол. журнал. — 2007. — № 4. — С. 37-43.
4. Неспецифическая интерстициальная пневмония и другие идиопатические интерстициальные пневмонии: классификация и критерии диагностики: по материалам статьи Анна-Луиз А. Катценстайн, Джеффри Л. Майерс (США, The American journal of surgical pathology) // Международный мед. журнал. — 2003. — Т. 6, № 3. — С. 244-246.
5. Попова Е.Н. Идиопатические интерстициальные пневмонии: клиника, диагностика, лечение // Лечащий врач. — 2005. — № 9.
6. Попова Е.Н., Болевич С.Б. Флуимицил при идиопатических интерстициальных пневмониях: эффективны ли высокие дозы? // Атмосфера. Пульмонология и аллергология. — 2006. — № 3. — С. 23-26.
7. Ребров А.П., Пономарева Е.Ю., Чеснокова Е.В. Идиопатический фиброзирующий альвеолит в практике терапевта // Клиническая медицина. — 2002. — № 9. — С. 63-65.
8. Фещенко Ю.И., Гаврисюк В.К., Моногарова Н.Е., Ячник А.И. Идиопатический фиброзирующий альвеолит как одна из форм идиопатических интерстициальных пневмоний // Укр. пульмонол. журнал. — 2004. — № 4. — С. 5-11.
9. Фещенко Ю.И., Гаврисюк В.К., Моногарова Н.Е. Без альтернативы. Лечение идиопатического фиброзирующего альвеолита // Ліки України. — 2005. — № 7, 8. — С. 71-73.
10. Фещенко Ю.И., Гаврисюк В.К., Моногарова Н.Е. Идиопатические интерстициальные пневмонии: классификация, дифференциальная диагностика // Укр. пульмонол. журнал. — 2007. — № 2. — С. 5-11.
11. Фещенко Ю.И., Гаврисюк В.К., Моногарова Н.Е. Идиопатические интерстициальные пневмонии // Therapia. — 2008. — № 1. — С. 34-40.
12. Чикина С.Ю. Применение Флуимицила в респираторной медицине // Атмосфера. Пульмонология и аллергология. — 2005. — № 3. — С. 36-40.
13. Чучалин А.Г. Идиопатический легочный фиброз // Терапевт. архив. — 2000. — № 3. — С. 5-12.
14. Шмелев Е.И. Идиопатический фиброзирующий альвеолит // Атмосфера. Пульмонология и аллергология. — 2004. — № 1. — С. 3-8.
15. American Thoracic Society, European Respiratory Society. Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement // Am. J. Respir. Crit. Care Med. — 2000. — Vol. 161. — P. 646-664.
16. American Thoracic Society / European Respiratory Society. International Multidisciplinary Consensus on the Idiopathic Interstitial Pneumonias // Am. J. Respir. Crit. Care Med. — 2002. — Vol. 165. — P. 277-304.
17. Brown K.K. Current management of idiopathic pulmonary fibrosis and predictors of outcome // New approaches to managing idiopathic pulmonary fibrosis / King T.E., ed. — American Thoracic Society, 2000. — C. 21-26.
18. Chan T.Y., Hansell D.M., Rubens M.B. Cryptogenic fibrosing alveolitis and the fibrosing alveolitis of systemic sclerosis: morphological differences on computed tomografic scans // Thorax. — 1997. — Vol. 52. — P. 265-270.
19. Coultas D.B., Zumwalt R.E., Blak W.C. The epidemiology of interstitial lung diseases // Am. J. Respir. Crit. Care Med. — 1994. — Vol. 150. — P. 967-972.
20. Daniil Z.D., Gilchrist P.C., Nichol-son A.G. A histologic pattern of nonspecific interstitial pneumonia is associated with a better prognosis than usual interstitial pneumonia in patients with cryptogenic fibrosing alveolitis // Am. J. Crit. Care Med. — 1999. — Vol. 160. — P. 899-905.
21. Dempsey O.J. Clinical review: idiopathic pulmonary fibrosis — past, present and furture // Respiratory medicine. — 2006. — Vol. 100. — P. 1871-1885.
22. Gribbin J., Hubbard R.B., Le Jeune I. et al. Incidence and mortality of idiopathic pulmonary fibrosis and sarcoidosis in the UK // Thorax. — 2006. — Vol. 61. — P. 980-985.
23. Hartman T., Primack S., Swensen S. Disease progression in usual interstitial pneumonia compared with desquamative interstitial pneumonia assessment with serial CT // Chest. — 1996. — Vol. 110. — P. 378-382.
24. Heyneman L. E., Ward S., Lynch D.A. et al. Respiratory bronchiolitis, respiratory bronchiolitis-associated interstitial lung disease, and desquamative interstitial pneumonia: different entities or part of the same disease process? // Am. J. Roentgenol. — 1999. — Vol. 173. — P. 1617 — 1622.
25. Hodgson U., Laitinen T., Tukiai- nen P. Nationwide prevalence of sporadic and familial idiopathic pulmonary fibrosis: evidence of founder effect among multiplex families in Finland // Thorax. — 2002. — Vol. 57, № 4. — P. 338-342.
26. Katzenstein A.-L.A., Myers J.L., Mazur M.T. Acute interstitial pneumonia. A clinicopathologic, ultrastructural, and cell kinetic study // Am. J. Surg. Pathol. — 1986. — Vol. 10. — P. 256-267.
27. King T.E., Mortenson R.L. Cryptogenic organizing pneumonitis. The North American experience // Chest. — 1992. — Vol. 102. — P. 8S-13S.
28. Leslie K.O. The pathology of idiopathic pulmonary fibrosis // New approaches to managing idiopathic pulmonary fibrosis / King T.E., ed. — American Thoracic Society, 2000. — C. 8-13.
29. Martinez F.J. Idiopathic interstitial pneumonias. Usual interstitial pneumonia versus nonspecific interstitial pneumonia // Proc. Am. Thorac. Soc. — 2006. — Vol. 3. — P. 81-95.
30. Muller N.L., Guerry-Force M.L., Staples C.A. et al. Differential diagnosis of bronchiolitis obliterans with organizing pneumonia and usual interstitial pneumonia: clinical, functional, and radiologic findings // Radiology. — 1987. — Vol. 162. — P. 151-156.
31. Nishimura K., Kitaichi M., Izumi T. Usual interstitial pneumonia: histologic correlation with high-resolution CT // Radiology. — 1992. — Vol. 182. — P. 337-342.
32. Olson J., Colby T.V., Elliott C.G. Hamman-Rich syndrome revisited // Mayo Clin. Proc. — 1990. — Vol. 65. — P. 1538-1548.
33. Quigley M., Hansell D.M., Nicholson A.G. Interstitial lung disease — the new synergy between radiology and pathology // Histopathology. — 2006. — Vol. 49. — P. 334-342.
34. Schwartz D.A. Epidemiology, morbidity, mortality, and familial distribution of idiopathic pulmonary fibrosis // New approaches to managing idiopathic pulmonary fibrosis / King T.E., ed. — American Thoracic Society, 2000. — C. 1-7.
35. Rughu G., Weycker D., Edelsberg J. et al. Incidence and prevalence of idiopathic pulmonary fibrosis // Amer. J. Resp. Crit. Care Med. — 2006. — Vol. 174. — P. 810-816.
36. Thabut G., Fourner M., Collard H.R., Brown K.K. Prognosis in idiopathic pulmonary fibrosis // Am. J. Respir. Crit. Care Med. — 2004. — Vol. 169, № 9. — P. 1075-1076.
39. Travis W.D., Matsui K., Moss J. Idiopathic nonspecific interstitial pneumonia: prognostic significance of cellular and librosing patterns: survival comparison with usual interstitial pneumonia and desquamative interstitial pneumo- nia // Am. J. Surg. Pathol. — 2000. — Vol. 24. — P. 19-33.
40. Von Plessen C., Grinde O., Gul-svik A. Incidence and prevalence of cryptogenic fibrosing alveolitis in a Norwegian community // Respiratory Medicine. — 2003. — Vol. 97, № 4. — P. 428-435.