
Газета «Новости медицины и фармации» Антимикробная терапия (343) 2010 (тематический номер)
Вернуться к номеру
Карбапенемы в современной клинической практике
Авторы: В.Б. Белобородов, д.м.н., профессор, К.П. Грувер, Кафедра инфекционных болезней РМАПО, г. Москва
Версия для печати
Резистентность бактерий представляет собой серьезную проблему антибактериальной терапии и в этом плане может иметь тяжелые социальные последствия. По сообщению агентства Рейтер, в 2004 году в США погибло около 70 тыс. пациентов с нозокомиальными инфекциями, причем у половины из них инфекции были вызваны флорой, резистентной к антибиотикам, которые обычно применяются для лечения таких инфекций. Опубликованы данные о более высокой летальности пациентов с инфекциями, вызванными резистентной флорой [1, 2]. Имеются сведения о дополнительных затратах системы здравоохранения, связанных с резистентностью нозокомиальной флоры, которые, по некоторым оценкам, составляют от 100 миллионов до 30 миллиардов долларов в год [3].
Основными механизмами резистентности микроорганизмов являются продукция ферментов, которые инактивируют антибиотики; нарушение или изменение структуры рецепторов, с которыми необходимо связаться антибиотикам для подавления бактериального роста; снижение концентрации антибиотиков внутри бактерий, связанное с невозможностью их попадания внутрь бактериальных клеток из-за нарушения проницаемости внешней оболочки или активного выведения с помощью специальных насосов.
Резистентность к антибиотикам наблюдается повсеместно и имеет неблагоприятную тенденцию к повышению. К настоящему времени, кроме резистентности к определенному препарату или группе препаратов, выделяют полирезистентные бактерии, т.е. резистентные к основным группам антибактериальных препаратов (β-лактамам, аминогликозидам, фторхинолонам), и панрезистентные, против которых, согласно данным микробиологических исследований, не имеется активных антибиотиков.
История создания антибактериальных препаратов была непосредственно связана с решением определенных клинических задач: поиском препаратов с высокой природной активностью для подавления стрептококков (пенициллин, ампициллин), стафилококков (оксациллин), грамотрицательной флоры (аминогликозиды); преодолением побочных эффектов (аллергия к природным пенициллинам); повышением пенетрации антибиотиков в ткани и клетки (макролиды, фторхинолоны). Однако применение антибиотиков привело к активации процессов защиты микрофлоры от них. Поэтому при разработке препаратов, которые в настоящее время широко применяются в клинике, стала актуальной задача преодоления природной и приобретенной резистентности нозокомиальной флоры. Наиболее яркими представителями этой относительно новой генерации препаратов являются карбапенемы.
Разработка карбапенемов и их структурно- функциональные особенности
Подобно пенициллинам и цефалоспоринам карбапенемы имеют природный источник. Первый карбапенем — тиенамицин является продуктом Streptomyces cattleya. Основной структурой тиенамицина и последующих карбапенемов, подобно пенициллинам, является пятичленное β-лактамное кольцо. Химической особенностью карбапенемов, отличающей их от пенициллинов, является замена углерода азотом в 1-й позиции и наличие двойных связей между 2 и 3 атомами углерода, высокая устойчивость к гидролизу β-лактамного кольца в 6-й позиции и наличие тиогруппы во 2-й позиции пятичленного кольца. Считается, что последнее из перечисленных отличий связано с повышенной антисинегнойной активностью карбапенемов.
Первый из карбапенемов — имипенем появился в клинической практике в 1986 году. Для повышения стабильности этого препарата против почечной дигидропептидазы-1 имипенем стали комбинировать с ингибитором этого фермента — циластатином, что существенно улучшило его фармакокинетику в почках.
Меропенем появился в клинической практике в 1996 году. Основным химическим отличием от имипенема было наличие трансгидроксиэтильной группы в 6-м положении, которая определяла стабильность препарата к действию различных β-лактамаз, уникальность микробиологических и фармакологических характеристик. Появление боковой диметилкарбамилпирролидинтиогруппы во 2-м положении пятичленного кольца резко повысило активность препарата против Pseudomonas aeruginosa и других важнейших грамотрицательных бактерий. Метильная группа в 1-й позиции создала стабильность препарата к действию почечной дигидропептидазы-1, что позволило использовать препарат без циластатина.
Эртапенем стал третьим препаратом в линейке карбапенемов в 2001 году. Подобно меропенему, он стабилен к почечной дигидропептидазе-1 и различным β-лактамазам. Химическим отличием этого препарата стало замещение метильной группы остатком бензойной кислоты во 2-й позиции пятичленного кольца, что резко повысило его связывание с белками плазмы. Этот показатель достигает 95 %, у имипенема — 20 % и 2 % — у меропенема. В результате этого увеличился период полувыведения препарата из плазмы, появилась возможность его введения 1 раз в сутки. Модификация химической структуры оказала негативное влияние на его активность в отношении неферментирующих грамотрицательных бактерий, таких как Pseudomonas aeruginosa и Acinetobacter baumannii [4–6]. В отношении Psedomonas aeruginosa предполагается, что существенное изменение заряда, увеличение молекулярного веса и липофильности нарушило пенетрацию эртапенема через мембранный пориновый канал (OprD), который является важнейшим порталом для пенетрации карбапенемов [7, 8].
В 2010 году появился новый карбапенем — дорипенем. Его химическая структура напоминает меропенем и эртапенем, отличается наличием сульфаммониламинометилпирролидинтиогруппы во 2-й позиции пятичленного кольца. Это изменение привело к повышению активности против Staphylococcus aureus, при этом активность против грамположительной флоры существенно не изменилась по сравнению с меропенемом [4–6, 9].
Механизм действия и значение пенициллинсвязывающих белков
Карбапенемы, как и другие β-лактамные антибиотики, являются бактерицидными ингибиторами синтеза клеточной стенки благодаря их связыванию с пенициллинсвязывающими белками (ПСБ). ПСБ — это цитоплазматические белки клеточной стенки, завершающие синтез пептидогликана — скелета клеточной стенки. Карбапенемы связываются со всеми основными ПСБ грамотрицательных бактерий. Основным отличием связывания с ПСБ карбапенемов и других β-лактамов является высокая аффинность к ПСБ-1а и -1b Pseudomonas aeruginosa и E.coli, что приводит к быстрому киллингу бактерий, увеличивает количество погибших бактерий. Среди карбапенемов, в свою очередь, имеются различия в аффинности к ПСБ-2 и -3 грамотрицательных бактерий. Имипенем имеет большее сродство к ПСБ-2 по сравнению с ПСБ-3. Это приводит к тому, что до возникновения лизиса бактерии приобретают сферическую или эллипсовидную форму. Однако аффинность к ПСБ-2 и -3 Pseudomonas aeruginosa одинаковая. Аффинность меропенема и эртапенема к ПСБ-2 и -3 E.coli значительно выше, чем у имипенема. Точно так же аффинность к ПСБ-2 Pseudomonas aeruginosa у меропенема выше, чем у имипенема, однако в отношении ПСБ-3 она выше в 3–10 раз. Аффинность к ПСБ-2, -3 у меропенема и дорипенема одинаковая [10, 11]. При этом имеются индивидуальные различия микробных штаммов в аффинности ПСБ к различным карбапенемам.
Фармакодинамические особенности карбапенемов
В большей степени зависят от кратности введения препаратов, чем от концентрации в крови, что отличает их от аминогликозидов и фторхинолонов, эффективность которых напрямую связана с концентрацией препарата в плазме. Максимальный бактерицидный эффект карбапенемов наблюдается при достижении концентрации в плазме, превышающей минимальную подавляющую концентрацию (МПК) в 4 раза [12]. В отличие от карбапенемов эффективность аминогликозидов и фторхинолонов возрастает пропорционально их концентрации в плазме и может быть ограничена только максимально разрешенной разовой дозой препарата [13].
Важнейшим фармакодинамическим показателем карбапенемов является отношение времени, когда концентрация препарата превышает МПК, ко времени между введениями препарата. Этот показатель выражается в процентах (T > МПК %). Теоретически идеально было бы поддерживать концентрацию карбапенема все 100 % интервала между введениями препарата. Однако это не обязательно для достижения оптимального клинического результата [14–16]. Более того, этот интервал является разным у различных β-лактамных антибиотиков. Для достижения бактериостатического эффекта антибиотика необходим показатель 30–40 % для пенициллинов и цефалоспоринов и 20 % — для карбапенемов. Для достижения максимального бактерицидного эффекта необходимо достижение показателя 60–70 % для цефалоспоринов, 50 % — для пенициллинов и 40 % — для карбапенемов [13, 17]. Несмотря на то что пенициллины, цефалоспорины и карбапенемы убивают бактерии с помощью одного механизма, различия в показателях T > МПК отражают различия в быстроте киллинга, который наименее быстрый у цефалоспоринов и наиболее быстрый — у карбапенемов [13]. Молекулярными причинами различия этого процесса у цефалоспоринов и карбапенемов может быть различная аффинность этих препаратов к ПСБ-1а и -1b.
Другой важной характеристикой этих препаратов является продолжительность постантибиотического эффекта (ПАЭ). ПАЭ — это эффект препарата, который продолжается после его удаления из системы. Среди β-лактамов ПАЭ наиболее часто наблюдается у карбапенемов. ПАЭ имипенема против некоторых микробов, включая P. aeruginosa, продолжается 1–4,6 часа [18–21]. Необходимо отметить, что этот показатель может существенно варьировать среди штаммов, принадлежащих к одному роду. У меропенема ПАЭ подобен имипенему [22]. Продолжительность ПАЭ эртапенема в отношении грамположительных бактерий составляет 1,4–2,6 часа. У дорипенема ПАЭ против S.aureus, K.pneumoniae, E.coli и P.aeruginosa наблюдался около 2 часов, причем только в отношении штаммов S.aureus и P.aeruginosa [23].
Спектр активности и клиническая эффективность
Карбапенемы имеют наиболее широкий спектр активности среди всех антибактериальных препаратов. Они активны против грамположительных и грамотрицательных микробов, включая аэробов и анаэробов. Показатель МПК50 позволяет оценить их природную активность и резистентность, по этому показателю они сходны с фторхинолонами и аминогликозидами. У некоторых бактерий отсутствует природная чувствительность к карбапенемам, например у S.maltophila, B.cepacia, E.faecium и резистентных к метициллину стафилококков [4, 5, 9, 24–26]. Имеются определенные различия между карбапенемами по природной активности, что может быть связано с нарушением пенетрации препаратов через клеточную мембрану и активности эффлюксных насосов. Данные по сравнительной активности всех 4 препаратов в отношении одних и тех же клинических штаммов микробов очень ограниченны. Однако имеются экспериментальные данные глобальных сравнительных исследований активности этих препаратов, которые также не являются исчерпывающими [4, 6]. Например, в одном из них нет сравнительной оценки определенных значений МПК: минимальная концентрация для дорипенема и меропенема составила 0,008 мкг/мл, для эртапенема — 0,06 мкг/мл, а для имипенема — 0,5 мкг/мл, поэтому у 3023 штаммов E.coli сравнение МПК90 оказалось возможным только при указанных выше показателях. Тем не менее имеются данные прямого сравнения МПК дорипенема, меропенема и имипенема в отношении энтеробактерий, P.aeruginosa, Haemophylus influenza и Bordetella pertussis, которые указывают на их сходную природную активность по показателю МПК50, который был аналогичным или отличался на одно-двукратное разведение [27, 28]. Только в отношении Proteus mirabilis активность меропенема была в 4 раза выше, чем активность дорипенема, и оба препарата оказались достоверно активнее имипенема, эти же тенденции сохранились и в отношении МПК90. Все три препарата оказались одинаково активными против чувствительных и резистентных к пенициллину S.pneumoniae. Резистентность, связанная с модификацией пенициллинсвязывающих белков, оказывала достоверное влияние на активность карбапенемов: МПК50 и МПК90 резистентных к пенициллину штаммов оказались в 32–64 раза выше, чем у чувствительных, при этом МПК90 оставалась ниже 1 мкг/мл. Дорипенем имел сходную с имипенемом активность против S.aureus и E.faecalis. Против чувствительных к цефтазидиму энтеробактерий, которые не продуцировали β-лактамаз расширенного спектра (БЛРС), активность эртапенема, меропенема и дорипенема была равной и превосходила таковую имипенема. Однако активность эртапенема была существенно ниже против неферментирующей грамотрицательной флоры (P.aeruginosa, A.baumannii) [4–6]. В отношении S.pneumoniae, S.aureus, S.epidermidis и E.faecalis активность карбапенемов была примерно одинаковой, включая эртапенем. В отношении грамположительных и грамотрицательных анаэробов активность карбапенемов также была одинаковой с МПК50 1 мкг/мл и ниже.
Карбапенемы и механизмы резистентности
Резистентность к β-лактамам имеется у грамотрицательных и грамположительных микроорганизмов. У грамположительных бактерий не имеется механизмов резистентности, связанных с изменением свойств внешней мембраны, или ферментов, способных разрушать карбапенемы. Появление резистентности грамположительных бактерий связано с изменением пенициллинсвязывающих белков (ПСБ), например с появлением ПСБ-2а с низкой аффинностью ко всем β-лактамам у резистентных к метициллину S.aureus (MRSA). У грамотрицательных бактерий наличие внешней мембраны и различных β-лактамаз приводило к появлению резистентности, связанной с продукцией инактивирующих ферментов (β-лактамаз), нарушением структуры ПСБ, снижением накопления препарата в перипластическом пространстве из-за снижения проницаемости белков-поринов внешней мембраны или эффлюксных насосов, выводящих различные антибиотики из микробной клетки. Из них наибольшее значение имеет продукция β-лактамаз и снижение клеточной проницаемости.
Бета-лактамазы расширенного спектра и класса AmpC
Продукция β-лактамаз является наиболее частым механизмом резистентности грамотрицательных бактерий. Расположение гидроэтилгруппы в положении 6 определяет высокую стабильность карбапенемов по сравнению с цефалоспоринами и пенициллинами к гидролизу β-лактамазами [29, 30], в особенности цефалоспориназами (БЛРС и AmpC). Поэтому реальным отличием карбапенемов от других β-лактамных антибиотиков является именно стабильность к действию БЛРС и AmpC.
AmpC — цефалоспориназы с широким спектром активности, разрушающие пенициллины (в том числе защищенные) и большинство цефалоспоринов. Необходимым условием разрушения антибиотиков является высокий уровень продукции этого фермента микробом. У P.aeruginosa и многих энтеробактерий (E.coli, K.pneumoniae) в хромосомах содержится информация о синтезе AmpC, однако синтез начинается при определенных условиях — при контакте с антибиотиком. Такой характер образования и выделения фермента называется индуцибельным. Однако при наличии врожденной предрасположенности к гиперпродукции фермента в результате мутации может происходить его депрессия [31]. Цефалоспориназы AmpC имеются на плазмидах некоторых энтеробактерий, наиболее часто они встречаются у K.pneumoniae и E.coli [32–34]. Некоторые передающиеся плазмидами AmpC могут иметь индуцибельный фенотип. Вне зависимости от того, является ли AmpC хромосомной или плазмидной, ее гиперпродукция у энтеробактерий и P.aeruginosa приводит к резистентности почти ко всем β-лактамам. Тем не менее многие энтеробактерии — гиперпродуценты AmpC остаются чувствительными к цефепиму и карбапенемам, а большинство P.aeruginosa — гиперпродуцентов AmpC оказываются чувствительными к имипенему, меропенему и дорипенему.
Продукция БЛРС является вторым механизмом резистентности к β-лактамам. Продукция этих ферментов приводит к резистентности к пенициллинам и цефалоспоринам [25, 36]. Источником этих ферментов для энтеробактерий оказалась Kluyvera spp. [37]. Необходимо отметить, что этот тип β-лактамаз может быть подавлен ингибиторами β-лактамаз (сульбактам, тазобактам, клавулановая кислота), поэтому защищенные пенициллины и цефалоспорины могут сохранять свою активность в отношении продуцентов БЛРС. Однако карбапенемы считаются препаратами выбора для лечения инфекций, вызванных энтеробактериями — продуцентами БЛРС [38]. Показано, что E.coli и K.pneumoniae остаются чувствительными ко всем карбапенемам, за исключением эртапенема, и МПК90 достоверно не изменяется. МПК90 эртапенема у продуцентов БЛРС оказывается примерно в 4 раза выше, чем у «диких» штаммов [39].
Карбапенемазы
Кроме БЛРС и AmpC, некоторые бактерии имеют ферменты (карбапенемазы), информация о которых кодирована на хромосоме или плазмидах [40]. Такие ферменты способны продуцировать некоторые энтеробактерии, P.aeruginosa и Acinetobacter spp. Карбапенемазы представляют сложную проблему для лечения тяжелых инфекций карбапенемами, однако прямой корреляции между продукцией карбапенемаз и резистентностью к карбапенемам выявить не удалось. Одним из объяснений этого факта является различие гидролитической активности карбапенемаз в отношении различных субстратов, каковыми являются различные препараты карбапенемов [41]. Другими причинами могут быть одновременное снижение пенетрации через бактериальную стенку (изменение структуры пориновых белков) или недоступность целевых пенициллинсвязывающих белков (наличие карбапенемаз в перипластическом пространстве). При наличии продукции карбапенемаз в клинических ситуациях не следует применять карбапенемы для лечения инфекций, вызванных такими микробами.
Резистентность, связанная с поринами
Снижение пенетрации внутрь бактериальной клетки является одним из механизмов резистентности к карбапенемам у энтеробактерий [41–45, 58]. Наиболее хорошо изучена резистентность P.aeruginosa, связанная с изменением структуры порина OprD, который осуществляет пассивный захват основных аминокислот и коротких пептидов, но также служит каналом для карбапенемов [46, 47]. Именно этот механизм резистентности характерен для карбапенемов и не влияет на чувствительность к другим β-лактамным АБ. У P.aeruginosa этот механизм связан с рядом генетических механизмов и приводит к повышению МПК имипенема в 4–16 раз, меропенема — в 4–32 раза, дорипенема — в 8–32 раза [48–51]. Несмотря на кажущееся преимущество имипенема, его МПК становится выше уровня, который рассматривается как чувствительный (4 мкг/мл), а МПК дорипенема и меропенема остаются ниже 4 мкг/мл [52].
Резистентность P.aeruginosa, связанная с эффлюксом
У потенциально резистентных P.aeruginosa в хромосоме имеются гены, кодирующие информацию о нескольких эффлюксных насосах, выводящих из клетки различные антибиотики. Наиболее изученными являются Mex-OprM, MexCD-OprJ, MexEF-OprN и MexXY. Эти насосы способны выкачивать из цитоплазмы и перипластического пространства клетки различные препараты. В результате изучения этих насосов открылись перспективы разработки новых антибактериальных препаратов, способных контролировать процесс их работы. С учетом этого стала понятной необходимость отдельного рассмотрения их роли в резистентности к имипенему, меропенему и дорипенему у P.aeruginosa.
Точно не установлены насосы, выводящие имипенем. Однако показано, что при высокой экспрессии двух эффлюксных насосов (MexCD-OprJ и MexEF-OprN) происходит значительное снижение чувствительности P.aeruginosa к имипенему. Показано, что этот механизм не связан с комбинацией β-лактамазной активности AmpC и OprD [53]. В то же время высокая экспрессия MexCD-OprJ и MexEF-OprN приводит к достоверному снижению чувствительности к имипенему за счет снижения экспрессии OprD [50, 54, 55].
В отличие от имипенема меропенем является подходящим субстратом для эффлюксных насосов: показано, что он выводится из клеток с помощью MexAB-OprM, MexCD-OprJ и MexEF-OprN [57]. По данным других исследований, только гиперпродукция MexAB-OprM определяет резистентность к меропенему [8]. Влиянием этого механизма объясняется различие в резистентности к имипенему и меропенему штаммов P.aeruginosa, имеющих такие насосы. Важно отметить, что повышенная продукция MexAB-OprM не обязательно приводит к подъему МПК выше уровня чувствительности [56], однако свидетельствует о вероятном взаимодействии этого механизма с другими (например, резистентности, связанной с OprD) и поэтому имеет важное клиническое значение. В отношении дорипенема показано, что он является субстратом для MexAB-OprM, MexCD-OprJ и MexEF-OprN эффлюксных насосов, более подробных сведений в литературе нет [57]. Таким образом, взаимодействие механизмов, связанных с выведением, нарушением проницаемости, β-лактамазной активности и доступности ПСБ приводит к клинически значимой резистентности к карбапенемам.
Дозирование и клиническая фармакокинетика
Все карбапенемы являются водорастворимыми веществами и вводятся внутривенно или внутримышечно из-за низкого всасывания из желудочно-кишечного тракта. Основные дозировки препаратов представлены в табл. 1.
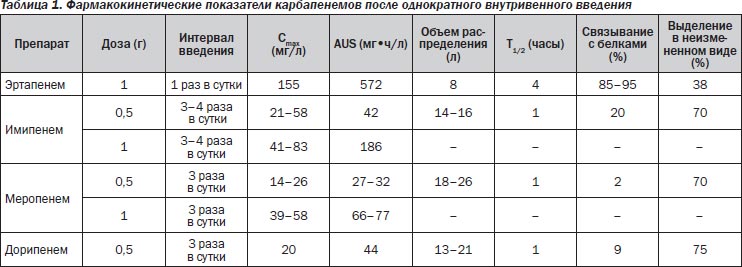
Величина связывания с белками является важным показателем фармакокинетики и антибактериальной активности препаратов. Фармакодинамический анализ антибактериальных препаратов требует учета связывания с белками и обсуждения кинетики именно «свободного» препарата. Как показано в табл. 1, связывание с белками имипенема (20 %), дорипенема (8 %) и меропенема (3 %) существенно различается [57, 58]. Изменение структуры эртапенема существенно повысило дозозависимое связывание с белками: до 95 % при концентрации в плазме ниже 100 мг/л и 85 % — выше 300 мг/л. Высокая связь с белками приводит к более продолжительному выведению: период полувыведения эртапенема составляет 4 часа по сравнению с 1 часом для других карбапенемов. Фармакокинетический профиль «свободного» препарата после введения дозы 500 мг показывает его эквивалентность у имипенема, меропенема и эртапенема. При этом преимущественно почечный клиренс препарата наблюдается у имипенема, меропенема и дорипенема.
Из-за продолжительного периода полувыведения эртапенем является единственным карбапенемом, который вводится 1 раз в сутки (500 мг или 1 г) [58]. Меропенем вводится по 500 мг или 1 г через 8 часов, а имипенем по 500 мг или 1 г через 6–8 часов. Снижение почечного клиренса требует снижения дозировки препаратов, однако при применении эртапенема этот клиренс должен быть ниже 30 мл/мин, при применении меропенема — ниже 51 мл/мин. Судорожный потенциал имипенема требует особого внимания при выборе дозировки препарата с учетом функции почек и массы тела. Снижение дозировки имипенема должно начинаться после снижения клиренса ниже 70 мл/мин и у пациентов с массой тела менее 70 кг.
Как было указано ранее, эффективность карбапенемов зависит от продолжительности интервалов между введениями препарата, когда его концентрация выше МПК. Оптимизация фармакодинамических показателей может быть достигнута с помощью введения более высокой дозы, укорочения периода между введениями и увеличения продолжительности инфузии препарата [59]. Наиболее привлекательным методом является увеличение продолжительности инфузии, т.к. это позволяет оптимизировать фармакодинамические показатели без существенного увеличения экономических затрат. Однако продолжительность инфузии лимитирована стабильностью препарата в растворе: меропенем и имипенем при комнатной температуре должны быть введены в течение 3 часов; стабильность дорипенема достигает 12 часов [58, 60]. В настоящее время продолжительная инфузия карбапенемов может рассматриваться в отношении меропенема и дорипенема. Однако максимально разрешенной дозировкой меропенема являются 6 г препарата в сутки, а дорипенема — 1,5 г/сут. Для оптимизации фармакодинамических показателей необходимо применение максимальной дозы и продолжительной инфузии препарата. Фармакодинамическое моделирование показало, что применение меропенема в дозе 6 г в сутки и 3-часовой инфузии создает условия для подавления флоры, которая интерпретируется при микробиологическом тестировании, как резистентная (до 64 мкг/мл). Возможность применения дорипенема в таких ситуациях ограничивается его низкой разрешенной суточной дозой (1,5 г).
Карбапенемы и судороги
Все β-лактамы способны вызывать судороги, особенно при неправильном дозировании в условиях нарушения функции почек или низкой массе тела, определенной хронической патологии или повышенной судорожной активности [61–64]. Повышение судорожной активности было выявлено еще в процессе проведения III фазы клинического исследования имипенема, а позже — меропенема и эртапенема [42, 58, 65]. Различные механизмы могут приводить к возникновению судорог, однако для карбапенемов основным механизмом является подавление рецепторов GABAa [66, 67]. Показано, что боковая цепь во 2-м положении 5-членого кольца карбапенемов является ответственной за это осложнение. Причем при наиболее высокой концентрации (10 ммоль/л) имипенем подавляет 95 % GABAа рецепторов, связывающих 3Н-мусцимол, меропенем подавляет 49 %, а дорипенем — 10 % [68]. Этим механизмом объясняется возникновение судорог у 1,5–6 % пациентов, получавших имипенем [61, 69, 70]. При ретроспективном исследовании дозозависимого эффекта было показано значение низкой массы тела, сниженной функции почек, наличия судорог в анамнезе, наличия другой патологии ЦНС и высоких доз имипенема/циластатина, которые нужно рассматривать в качестве факторов риска возникновения судорог [61]. Избыточной дозой имипенема/циластатина является та, которая превышает рекомендуемую дневную дозу на 25 %, и обычная доза у пациентов с нарушением функции почек или сопутствующей патологией ЦНС. Тщательный контроль дозирования препарата позволил снизить частоту возникновения судорог до уровня, который наблюдается при применении меропенема и эртапенема (~0,5 %) [70–72].
Заключение
Карбапенемы в настоящее время остаются наиболее надежными препаратами для лечения нозокомиальных инфекций у тяжелых пациентов, особенно в случаях инфекций, вызванных резистентной флорой. С учетом современных тенденций роста и распространения резистентности нозокомиальной флоры карбапенемы являются основными препаратами для лечения инфекций, вызванных резистентными грамотрицательными микробами (энтеробактериями, P.aeruginosa, Acinetobacter spp.). Разрешенные суточные дозы и возможность продленной инфузии позволяют рассматривать меропенем как единственный препарат, фармакодинамика которого может быть оптимизирована для подавления флоры, которая с микробиологических позиций определяется резистентной к меропенему и другим карбапенемам.
Впервые опубликовано в «Русском медицинском журнале». — 2010. — том 18, № 17.
1. Chow J.W. et al. // Ann. Intern. Med. — 1999. — 115. — 585-590.
2. Holmberg S.D. et al. // Rev. Infect. Dis. — 1987. — 9. — 1065-1078.
3. Phelps C.E. // Med. Care. — 1989. — 27. — 193-203.
4. Firtsche T.R. et al. // Clin. Microbiol. Infect. — 2005. — 11. — 974-984.
5. Ge Y. et al. // Antimicrob. Agents Chemother. — 2004. — 48. — 1384-1396.
6. Jones R.N. et al. // J. Antimicrob. Chemother. — 2004. — 54. — 144-154.
7. Hammond M.L. // J. Antimicrob. Chemother. — 2004. — 53 (Suppl. 2). — ii7-ii9.
8. Kohler T.J. et al. // Antimicrob. Agents Chemother. — 1999. — 43. — 424-427.
9. Iso Y. et al. // J. Antibiot. — 1996. — 49. — 199-209.
10. Davis T.A. et al. // ICAAC. — 2006 (Abstract C1-0039).
11. Fujimura T. et.al. // Jpn. J. Chemother 2005. — 53 (Suppl. 1). — 56-69.
12. Craig W. // Diagn. Microbiol. Infect Dis. — 1995. — 22. — 89-96.
13. Craig W. // Clin. Infect. Dis. — 1998. — 26. — 1-12.
14. Craig W. // Scand. J. Infect. Dis. — 1991. — 74. — 63-70.
15. Wogelman D. et al. // J. Infect. Dis. — 1985. — 152. — 373-378.
16. Roosendaal R. et al. // J. Infect. Dis. — 1985. — 152. — 373-378
17. DeRyke C.A. et al. // Drug. — 2006. — 66. — 1-14.
18. Hanberger H. et al. // Eur. J. Clin Microbiol. Infect. Dis. — 1991. — 10. — 927-934.
19. Bustamante C.I. et al. // Antimicrob. Agents Chtmother. — 1984. — 26. — 678-683.
20. Gudmundsson S. et al. // J. Antimicrob. chemother. — 1986. — 18. — 67-73.
21. Nadler H.L. et al. // J. Antimicrob. chemother. — 1989. — 24 (Suppl. 1). — 225-231.
22. Odenholt I. // Expert Opin. Investig. Drugs. — 2001. — 10. — 1157-1166.
23. Totsuka K., Kikuchi K. // Jap. J. Chemother. — 2005. — 53 (Suppl.1). — 51-55.
24. Livermore D.M. et al. // J. Antimicrob. chemother. — 2003. — 52. — 331-344.
25. Pryka R.D., Haig G.M. // Ann. Pharmacother. — 1994. — 28. — 1045-1054.
26. Jones R.N. // Am J. Med. — 1985. — 78 (Suppl. 6A). — 22-32.
27. Brown S.D., Traczewski M.M. // J. Antimicrob. chemother. — 2005. — 55. — 944-949.
28. Tsuji et al. // Antimicrob. Agents Chemother. — 1998. — 42. — 94-99.
29. Cassidy P.J. // Dev. Ind. Microbiol. — 19881. — 22. — 181-209.
30. Miyashita K. et al. // Bioorg. Med. Chem. Lett. — 1996. — 6. — 319-322.
31. Hanson N.D., Sanders C.C. // Curr. Pharm. Des. — 1999. — 5. — 881-894.
32. Hanson N.D. // J Antimicrob. chemother. — 2003. — 52. — 2-4.
33. Perez F., Hanson N.D. // J. Antimicrob. chemother. — 2002. — 40. — 2153-2162.
34. Jacoby G.A. // Antimicrob. Agents Chemother. — 2006. — 50. — 1123-1129.
35. Bradford P.A. // Clin Microbiol. Rev. — 2001. — 14. — 933-951.
36. Jacoby G.A. // Eur J. Clin. Microbiol. Infect. Dis. — 1994. — 13 (Suppl. 1). — 2-11.
37. Bonnet R. // Antimicrob. Agents Chemother. — 2004. — 48. — 1-14.
38. Bradford P.A. et al. // Clin. Infect. Dis. — 2004. — 39. — 55-60.
39. Jones R.N. et al. // Diag. Microbiol. Infect. Dis. — 2005. — 52. — 71-74.
40. Bonfigio G. et al. // Expert Opin. Investig. Drugs. — 2002. — 11. — 529-544.
41. Livermore D.M. et al. // Antimicrob. Agents Chemother. — 2001. — 45. — 2831-2837.
42. Mushtag S. et al. // Antimicrob. Agents Chemother. — 2004. — 48. — 1313-1319.
43. Koh T.N. et al. // Antimicrob. Agents Chemother. — 2001. — 45. — 1939-1940.
44. Jacoby G.A. et al. // Antimicrob. Agents Chemother. — 2004. — 48. — 3203-3206.
45. Mertinez-Martinez L. et al. // Antimicrob. Agents Chemother. — 1999. — 43. — 1669-1673.
46. Trias J., Nikaido H. // Antimicrob. Agents Chemother. — 1990. — 34. — 52-57.
47. Trias J., Nikaido H.J. // Biol. Chem. — 1990. — 265. — 15680-15684.
48. Wolter D.J. et al. // FEMS Microbiol. Lett. — 2004. — 236. — 137-143.
49. Yoneyama H., Nakae T. // Antimicrob. Agents Chemother. — 1993. — 37. — 2385-2390.
50. Ochs M.M. et al. // Antimicrob. Agents Chemother. — 1999. — 43. — 1085-1090.
51. Sakyo S. et al. // J. Antibiol. — 2006. — 59. — 220-228.
52. Lister P. // Antimicrob. Agents Chemother. — 2005. — 49. — 4763-4766.
53. Fukuda H. et al. // Antimicrob. Agents Chemother. — 1995. — 39. — 790-792.
54. Lister P., Wilter D.J. // Clin/ Infect. Dis. — 2005. — 40. — S105-S114.
55. Masuda N. et al. // Antimicrob. Agents Chemother. — 1995. — 39. — 645-649.
56. Masuda N. et al. // Antimicrob. Agents Chemother. — 2000. — 44. — 3322-3327.
57. Physicians’ Desk Reference. — Thomson, 2005.
58. Mattoes H.M. et al. // Clin Ther. — 2004. — 26. — 1187-1198.
59. Psathas P. et al. // American Society of Health-System Pharmacists. — San Francisco, 2007. — Abst 57E.
60. Calandra G.B. et al. // Am J. Med. — 1988. — 84. — 911-918
61. De Sarro A. et al. // Neuropharmacology. — 1989. — 28. — 359-365.
62. Williams P.D. et al. // Antimicrob. Agents Chemother. — 1988. — 32. — 758-760.
63. Barrons R.W. et al. // Ann. Pharmacother. — 1992. — 26. — 26-29.
64. Lucasti C. et al. // Europ. Cong. Clin. Microbiol. Infect. Dis. — 2007. — Abstr. P834
65. Day L.P. et al. // Toxicol. Lett. — 1995. — 76. — 239-243.
66. Shimuda J. et al. // Drug Exp. Clin. Res. — 1992. — 18. — 377-381.
67. Horiuchi M. et al. // Toxicology. — 2006. — 222. — 114-124.
68. Job M.I., Dretler R.H. // Ann. Pharmacother. — 1990. — 24. — 467-469.
69. Pestotnik S.L. et al. // Ann. Pharmacother. — 1993. — 27. — 497-501.
70. Rodloff A.C. et al. // J. Antimicrob. Chemother. — 2006. — 58. — 916-929.
71. Kearing G.M., Perry C.M. // Drugs. — 2005. — 65. — 2151-2178.