
Международный неврологический журнал 1 (39) 2011
Вернуться к номеру
Синдром паркинсонизма у молодых женщин, страдающих дефицитом миелопероксидазы фагоцитов
Авторы: Казмирчук В.Е.1, Мальцев Д.В.1, Слободин Т.Н.2, Головченко Ю.И.2, 1Институт иммунологии и аллергологии Национального медицинского университета имени А.А. Богомольца, г. Киев, 2Кафедра неврологии НМАПО имени П.Л. Шупика, г. Киев
Рубрики: Неврология
Версия для печати
Впервые приведено описание историй болезни трех молодых женщин, страдающих дефицитом миелопероксидазы фагоцитов, у которых болезнь поначалу проявлялась в виде инфекционного синдрома, а в третьей части жизни развились проявления паркинсонизма. Эти наблюдения согласуются с сообщениями о повышенном риске нейродегенеративных осложнений и паркинсонизма, в частности, у пациентов с другими формами первичных иммунодефицитов, а именно при синдроме Чедиака — Хигаси, наследственном аутосомно-рецессивном заболевании, сходном по патогенезу с дефицитом миелопероксидазы, и Х-сцепленной агаммаглобулинемии (болезни Брутона), которая также имеет много общего с иммунодефицитами, в основе которых лежит фагоцитарная недостаточность, учитывая опсонизирующую активность антител. Это сообщение позволяет по-новому взглянуть на перспективы лечения экстрапирамидных расстройств при помощи средств современной иммунотерапии.
Дефицит миелопероксидазы, паркинсонизм, первичный иммунодефицит, нейродегенеративные болезни.
 Одним из знаменательных достижений современной нейроиммунологии является установление воспалительной природы нейродегенеративных процессов. Действительно, признаки воспаления обнаружены в зоне сенильных бляшек при болезни Альцгеймера [4], кортико-спинальных путей и передних рогов спинного мозга при боковом амиотрофическом склерозе [21], черной субстанции среднего мозга при болезни Паркинсона [5], гиппокампа и медиальных отделов височных долей при так называемой темпоральной мезиальной эпилепсии (англ. temporal mesial epilepsy) [59].
Одним из знаменательных достижений современной нейроиммунологии является установление воспалительной природы нейродегенеративных процессов. Действительно, признаки воспаления обнаружены в зоне сенильных бляшек при болезни Альцгеймера [4], кортико-спинальных путей и передних рогов спинного мозга при боковом амиотрофическом склерозе [21], черной субстанции среднего мозга при болезни Паркинсона [5], гиппокампа и медиальных отделов височных долей при так называемой темпоральной мезиальной эпилепсии (англ. temporal mesial epilepsy) [59].
Этим достижениям предшествовало изменение современных представлений о природе воспаления как такового, которое ранее рассматривалось исключительно как вазогенный процесс, проявлениями которого являются гиперемия, плазморрагия, диапедез и клеточная инфильтрация. Сегодня стало известно, что воспаление также может быть и тканевым, или локальным по своему происхождению, т.е. реализовываться благодаря не факторам сыворотки крови, а компонентам местного иммунитета того или иного компартмента человеческого организма. Признаками такого воспалительного процесса являются повышенная продукция провоспалительных цитокинов, активация оксирадикального стресса и изменение экспрессии ряда поверхностных молекул, в частности рецепторов к цитокинам, адгезионных и костимулирующих структур [59]. Именно такие признаки отмечаются в образцах ткани, полученных из очагов нейродегенерации.
Хотя доказательство роли воспаления при дистрофии является важным достижением, однако это открытие ставит на повестку дня большое количество принципиальных вопросов, ответы на которые все еще не получены. Во-первых, является ли воспаление причиной нейродегенерации или лишь вторичным процессом, развивающимся вследствие индукции нейродегенеративных изменений? В серии экспериментальных работ было убедительно продемонстрировано, что индукция воспаления в мозге лабораторных животных может приводить к развитию патоморфологических и клинических симптомов, аналогичных таковым при известных нейродегенеративных болезнях, в частности болезни Паркинсона, а использование противовоспалительных средств, например циклоспорина А [39, 40], может остановить процесс нейродегенерации. Однако мы до сих пор не располагаем достаточным объемом подобных доказательств, полученных при изучении людей, страдающих нейродегенеративными поражениями. Тем не менее результаты одного из последних исследований, свидетельствующие о протективном влиянии терапии ибупрофеном на риск развития болезни Паркинсона [12], позволяют сделать обоснованный вывод о том, что воспаление является принципиально важным компонентом патогенеза, а не только простым «свидетелем» нейродегенеративных изменений.
Во-вторых, не являются ли инфекционные агенты истинной причиной нейродегенерации, учитывая хорошо известный факт, что воспаление — универсальная реакция организма на микробное вторжение? Действительно, в этом направлении уже имеется ряд доказательств. Всегда бросался в глаза тот факт, что при болезни Альцгеймера избирательно поражаются именно те области мозга, к которым проявляют повышенный тропизм вирусы герпеса [17]. В серии исследований продемонстрировано наличие ДНК вируса простого герпеса 1-го типа (ВПГ-1) в биоптатах, полученных из зон сенильных бляшек у пациентов с болезнью Альцгеймера [25, 26, 62]. Более того, обнаружено, что наличие аллеля аполипопротеина e4 (АРоЕ e4) в большей мере предрасполагает к развитию болезни Альцгеймера у тех пациентов, у которых в паренхиме головного мозга имеется ВПГ-1, по сравнению с больными, которые являются только носителями аллеля АРоЕ e4 [3, 16]. Установлено также, что нейроинвазивность ВПГ-1 зависит от общего количества АРоЕ и особенно от наличия изоформы e4, что способствует проявлению латентности этого вируса в ткани головного мозга [8].
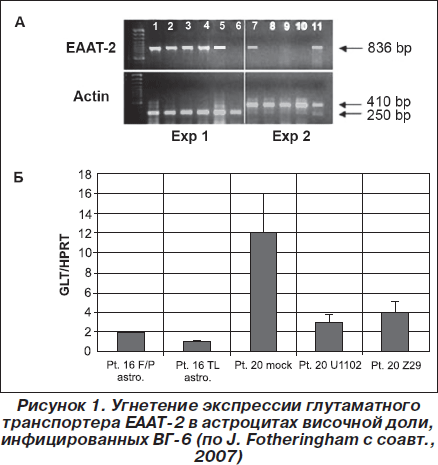
В то же время в серии недавних научных публикаций сообщается о наличии в биоптатах мозга, полученных от больных с височной мезиальной эпилепсией, еще одним нейродегенеративным заболеванием, характеризующимся прогрессирующим гиппокампальным склерозом, ДНК вируса герпеса 6-го типа (ВГ-6). При изучении на предмет наличия ДНК ВГ-6 образцов мозговой ткани от здоровых добровольцев, пациентов с другими нейродегенеративными болезнями или неокортикальной эпилепсией получены отрицательные результаты [18–20, 29, 55, 56]. Более того, S. Meeuwsen с соавт. в серии экпериментальных работ на клеточных культурах продемонстрировали потенциальный механизм проэпилептогенного эффекта ВГ-6, выявив усиленную продукцию ряда провоспалительных цитокинов глиального происхождения и связанное с этим изменение чувствительности нейронов к проэпилептогенным стимулам, ассоциированные с репродукцией этого вируса в клетках глии. Однако самыми важными являются результаты клинических исследований J.Fotheringham с соавт. (2007, 2008), выполненных на биоптатах гиппокампа, полученных от больных с височной мезиальной эпилепсией. Авторы не только показали присутствие ДНК ВГ-6, но и убедительно продемонстрировали механизм, при помощи которого персистирующий патоген оказывает проэпилептогенное воздействие, в частности снижение синтеза глутаматного транспортера ЕААТ-2 астроцитов, что коррелировало с повышением экспрессии гликопротеина ВГ-6 gp116/54/64 (рис. 1). Такие вирус-индуцированные изменения опосредуют уменьшение обратного захвата глутамата, что приводит к резкому возрастанию содержания этой возбуждающей аминокислоты во внеклеточном пространстве. Это способствует развитию глутаматергической эксайтотоксичности, обусловливающей прогрессирующий гиппокампальный склероз и реализацию проконвульсивных эффектов глутамата [19, 20].
Повышение частоты паркинсонизма в человеческой популяции после эпидемии энцефалита Экономо, наблюдавшееся в первой половине ХХ столетия, обосновывает гипотезу вирусного происхождения акинетико-ригидного синдрома, связанного с поражением черной субстанции среднего мозга. Более того, некоторые типичные симптомы болезни Паркинсона, например аносмия, которая является характерным проявлением дебюта болезни, могут быть ассоциированы с герпесвирусной нейроинфекцией, вызванной ВПГ-1, учитывая хорошо известный факт миграции этого вируса в центральной нервной системе (ЦНС) посредством обонятельных нервов и типичную зону латентности или репродукции этого патогена в клетках гиппокампа, где находится центральная часть обонятельного анализатора [3, 17].
Заслуживает внимания гипотеза ольфакторного патогенеза болезни Паркинсона [35]. Предполагается, что аномальная складчатая b-форма a-синуклеина, образуясь в эпителии обонятельных луковиц под влиянием факторов внешней среды, затем трансневрально распространяется в структуры мозга, имеющие связи с обонятельными луковицами. Подтверждением этому служат результаты экспериментальной ольфакторной инокуляции вируса герпеса 1-го типа и обезьяньего гепатита [35]. Стадийность распространения патологического процесса с постепенным вовлечением образований ствола, лимбической системы и коры описывается гипотезой Н. Braak [6]. Принимая во внимание прионную теорию S. Prusiner [50], автор предполагает, что по аналогии с прионными болезнями при болезни Паркинсона патогенный белок в виде b-формы a-синуклеина, становясь матрицей, приводит к своему повторению в уязвимых структурах мозга с последующей индукцией их гибели.
Симптомы экстрапирамидной недостаточности, сходные с таковыми при болезни Паркинсона, развиваются у некоторых ВИЧ-инфицированных пациентов, причем непосредственной причиной неврологических расстройств является токсоплазмоз мозга — типичная оппортунистическая инфекция, сопровождающаяся гранулематозным поражением ткани мозга и часто встречающаяся у иммуноскомпрометированных пациентов [9, 36].
В то же время существуют мнения, что паркинсонизм, имитирующий первичное нейродегенеративное заболевание, может быть обусловлен билатеральными грибковыми абсцессами в области стриатума, а терапия при помощи амфотерицина В может способствовать купированию симптомов имеющейся экстрапирамидной недостаточности [1].
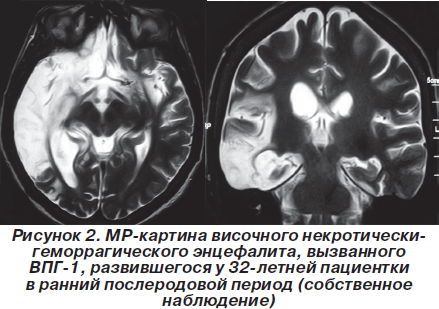
И, наконец, в-третьих, не являются ли болезни иммунной системы вовлеченными в патогенез нейродегенеративных процессов, учитывая повышенную склонность иммуноскомпрометированных пациентов к генерации хронических воспалительных процессов и индукции рецидивирующих инфекционных эпизодов? Сегодня описано 2 формы первичного иммунодефицита, проявляющиеся исключительно височным энцефалитом, вызванным ВПГ-1 (рис.2), в частности аутосомно-доминантный дефицит TLR-3 [63] и аутосомно-рецессивная недостаточность белка UNC93B— компонента внутриклеточного каскада, активируемого TLR-3 [10]. Поэтому важным является вопрос о возможной ассоциации между этими иммунодефицитами и риском развития болезни Альцгеймера в старости, учитывая известные данные о роли ВПГ-1 в развитии этого нейродегенеративного заболевания.
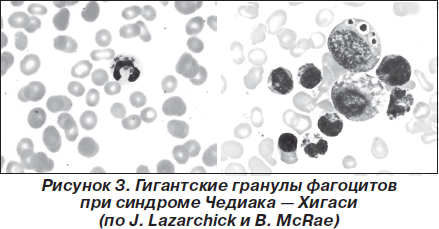
Кроме того, предположение о взаимосвязи между наследственными болезнями иммунной системы и неврологическими осложнениями подтверждают многочисленные сообщения о повышенной частоте нейродегенеративных заболеваний в третьей части жизни у пациентов с синдромом Чедиака — Хигаси— первичным иммунодефицитом с аутосомно-рецессивным типом наследования. В основе этой болезни лежит фагоцитарная недостаточность, связанная с мутацией в гене, отвечающем за регуляцию перемещения лизосом (англ. lysosomal trafficking regulator gene, ltr-gene), располагающемся в локусе 1q42–43 [54]. У таких больных не происходит надлежащего слияния фагосом с первичными лизосомами, что позволяет фагоцитированным микроорганизмам, в основном бактериальным и грибковым патогенам, избегать уничтожения при помощи микробицидных механизмов. Критериями диагностики синдрома Чедиака — Хигаси являются иммунная недостаточность, парциальный окулокутанный альбинизм, горизонтальный нистагм, крупные эозинофильные, пероксидазо-положительные тельца включений в гранулосодержащих клетках (рис. 3).
Отмечено, что примерно у 10 % пациентов, у которых отмечается легкая иммунная недостаточность, позволяющая избежать летального исхода от инфекционных осложнений в первые два десятилетия жизни, позже закономерно отмечается развитие нейродегенеративных болезней, в частности паркинсонизма, деменции, спиноцеребеллярной дистрофии и периферической нейропатии [57]. У этих пациентов отмечается аномальное распределение меланина в коже и клетках нервной системы, связанное с дисфункцией траспортных белков, кодируемых ltr-геном, что может объяснить сочетание симптомов альбинизма и паркинсонизма. Однако взаимосвязь между первичным иммунодефицитом и неврологическими осложнениями можно усмотреть еще и в том, что при синдроме Чедиака— Хигаси имеет место нарушение переработки экзогенных и эндогенных молекул не только в фагоцитах, но и в нейронах и клетках глии, так как продукты ltr-гена обслуживают широкий спектр клеток, не ограничиваясь циркулирующими моноцитами и нейтрофилами. Действительно, на сегодняшней день ведущей теорией, объясняющей патогенез нейродегенеративных болезней, является теория нарушенной аутофагии, т.е. несостоятельности системы утилизации некоторых эндогенных молекул, например бета-амилоида при болезни Альцгеймера [34] или альфа-синуклеина при паркинсонизме [49], что приводит к их избыточному накоплению в нервной ткани, запуску воспалительной реакции и индукции нейродегенерации. Так, можно обоснованно заключить, что фагоцитарная недостаточность, наблюдающаяся при синдроме Чедиака — Хигаcи, с одной стороны, обусловливает нарушение переработки экзогенных микробных молекул, что в клинике проявляется рецидивирующим инфекционным синдромом, симптомы которого возникают с самого раннего детства, а с другой стороны — дефектом обработки и утилизации эндогенно синтезированных структур, что опосредует развитие прогрессирующего нейродегенеративного процесса в третьей части жизни.
В частности, Е. Uyama c cоавт. (1991, 1994) описали клинический случай тяжелого паркинсонизма, проявляющегося почти полным обездвиживанием и резким усилением тонуса скелетных мышц по пластическому типу, который сопровождался атрофией зрительных нервов, обесцвечиванием радужки, горизонтальным нистагмом и наличием гигантских гранул в цитоплазме лейкоцитов крови, у 39-летней пациентки с синдромом Чедиака — Хигаси, первые симптомы экстрапирамидной недостаточности у которой возникли в возрасте 22 лет. МРТ демонстрировала атрофию височных долей полушарий и спинного мозга. При психоневрологическом исследовании были обнаружены признаки деменции [57, 58]. R.A. Hauser c соавт. (2000) доложили о случае прогрессирующего паркинсонизма у мужчины 29 лет с синдромом Чедиака — Хигаси, верифицированном на основании идентификации гигантских цитоплазматических гранул фагоцитов крови. У пациента отмечались тремор покоя и резкое усиление тонуса мышц с феноменом «зубчатого колеса» наряду с проявлениями гиперкинеза по типу дистонии, причем была зарегистрирована отчетливая положительная динамика со стороны тремора при использовании комбинированной терапии леводопой и амантадином [24]. L. Silveira-Moriyama с соавт. (2004) описали клинический случай прогрессирующего паркинсонизма у 24-летней женщины с верифицированным диагнозом синдрома Чедиака — Хигаcи и продемонстрировали эффективность терапии специфическими антипаркинсоническими препаратами [54]. С. Jacobi c cоавт. (2005) описали еще один случай паркинсонизма у пациента с указанным первичным иммунодефицитом и об-основали положение о пресинаптическом уровне расстройств дофаминергической системы в приведенном примере [27].
Интересно, что экстрапирамидные расстройства встречаются и при других первичных иммунодефицитах. В частности S. Papapetropoulos c соавт. (2007) описали прогрессирующий, фатальный синдром «паркинсонизм— дистония» с проявлениями деменции у молодого пациента с Х-сцепленной агаммаглобулинемией (болезнью Брутона), обусловленной мутацией гена тирозинкиназы В-лимфоцитов, причем больной на протяжении 14 лет получал заместительную терапию препаратами внутривенного иммуноглобулина, что позволяло успешно купировать кокковые инфекции дыхательной системы, которые обычно вызывают хроническую пневмонию у таких больных [47]. N.Quinn (1990) доложил о развитии псевдопаркинсонического тремора и дистрофической полиневропатии у пациента с дисиммуноглобулинемией [52]. J.T. Brookes (2007) с соавт. сообщают об ассоциации еще одной нейродегенеративной патологии, так называемого синдрома «глухота— дистония — неврит зрительного нерва» (англ. DDON— deafness-dystonia-optic neuronopathy), с болезнью Брутона [7]. Эти наблюдения указывают на то, что в патогенез некоторых нейродегенеративных расстройств, и в частности паркинсонизма, помимо фагоцитарной недостаточности может быть вовлечена гипоиммуноглобулинемия [64]. Как известно, антитела выполняют функцию опсонинов, т.е. облегчают фагоцитоз, что сближает между собой фагоцитарные нарушения и гипоиммуноглобулинемию. В то же время известно, что при гипоиммуноглобулинемии развиваются атипично протекающие энтеро- [51] и полиовирусные [37] нейроинфекции, поэтому инфекционный фактор также может рассматриваться как непосредственная причина нейродегенерации при болезни Брутона.
Мы приводим описание историй болезни 3 пациенток с паркинсонизмом, страдающих дефицитом миелопер-оксидазы фагоцитов, — распространенным первичным иммунодефицитом с аутосомно-рецессивным типом наследования, сходным по своей природе с синдромом Чедиака — Хигаси.
Как известно, миелопероксидаза является антимикробным белком, содержащимся в азурофильных гранулах нейтрофилов и лизосомах моноцитов и обеспечивающим образование из кислорода молекул перекиси водорода (Н2О2) и гипохлорит-аниона (НСlO–), которые наделены выраженными микробицидными свойствами [31]. Наследственный дефицит миелопероксидазы считается аутосомно-рецессивным расстройством, однако подчеркивается, что по типу наследования эта болезнь несколько отличается от классических рецессивных заболеваний тем, что недостаточность фагоцитоза отмечается не только у гомозигот, но и у многих гетерозигот, что связано с разными типами мутаций в гене миелопероксидазы, которые лежат в основе этой патологии [32]. Ген миелопероксидазы расположен в локусе 17q12–q21. Так, Nauseef et al., изучая генетические аномалии, вызывающие недостаточность миелопероксидазы, пришли к выводу, что эту болезнь могут обусловить снижение синтеза предшественника одного из пептидов, составляющих этот фермент, нарушение регуляции продукции обоих пептидов, а также аномальные постсинтетический процессинг и упаковка макромолекулы в азурофильных гранулах [41]. Одной из типичных причин дефицита миелопероксидазы среди населения США является миссенс-мутация arg569-to-trp с заменой аргинина на триптофан [42], хотя в Японии преобладает мутация R499C, при которой отмечается замена аргинина на цистеин [44, 48], что, по-видимому, определяет отличия в частоте встречаемости этой болезни в указанных странах [28].
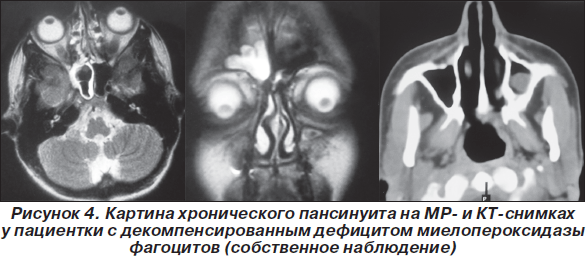
В детском и зрелом возрасте это заболевание проявляется преимущественно инфекционным синдромом с превалирующим поражением верхних дыхательных путей, придаточных пазух носа и кожи, вызванным условно-патогенной бактериальной или грибковой микрофлорой, особенно С.albicans [2], Е.сoli [22, 53], и, по-видимому, некоторыми видами стафило- и стрептококков [31]. Пансинуит является типичной клинической находкой у таких больных (рис. 4). Этим пациентам часто производится тонзилл- или аденоидэктомия в связи с часто рецидивирующими ангинами, создающими угрозу развития острой ревматической лихорадки, и хроническим аденоидитом, затрудняющим носовое дыхание.
Иногда инфекционный синдром осложняется аллергическими проявлениями, преимущественно атопическим дерматитом, ринитом или пищевой аллергией. В последнее время появились сообщения о повышенном риске развития миелопролиферативных неоплазий у пациентов с наследственным дефицитом миелопероксидазы [45, 61]. Тяжесть клинических проявлений этого иммунодефицита существенно варьирует от бессимптомных форм, при которых отсутствуют проявления инфекционного синдрома в связи с успешной реализацией компенсаторных механизмов, например, за счет активации НАДФ-оксидазы [46], до маргинальных клинических случаев, сопровождающихся потенциально летальными поражениями. Так, Kitahara et al. описали двух больных с дефицитом миелопероксидазы, которые страдали рецидивирующими стафилококковыми флегмонами и менингитами [31]. Р. Cech c cоавт. сообщают, что дефицит миелопероксидазы у пациента с сопутствующим сахарным диабетом привел к формированию множественных абсцессов в печени, вызванных C.albicans [11].
Больная К.О.В., 47 лет, в 43-летнем возрасте отметила первые симптомы болезни в виде тремора в пальцах левой руки, легкой неловкости и скованности в левой кисти, спустя 2 года присоединилась скованность в левой ноге, нарушилась походка. Учитывая наличие болевого синдрома в области левого плеча, а также периодически возникающие преходящие боли в крупных суставах, общую слабость, утомляемость, больная лечилась по поводу остеохондроза на протяжении 3 лет. Из анамнеза: частые обострения гайморита, других синуитов, упорно рецидивирующие герпетические высыпания на коже и слизистых оболочках. На момент осмотра: гипомия; гипокинезия и мышечная ригидность с преобладанием в левых конечностях; легкий непостоянный тремор покоя в пальцах левой кисти; постуральная неустойчивость (по Хен и Яру — 2,5ст.). Отмечает нарушение обоняния на протяжении как минимум 5 лет. Другой неврологической симптоматики не выявлено. Болезнь Паркинсона в семье отрицает.
Болеет хроническим ринофарингитом, тонзиллитом, аденоидитом, часто рецидивирующими ангинами, синуитами с преимущественным поражением верхнечелюстных пазух, упорно рецидивирующей молочницей; отмечались периоды субфебрилитета и лимфаденопатии неясной этиологии, имеет место склонность к частым ОРВИ с последующими бактериальными осложнениями.
Общий анализ крови (27.07.10): эр. — 4,6 х 1012/л; Нb— 141 г/л; ЦП — 0,97; тромбоциты — 363 ´ 109/л; лейкоциты— 8,8 ´ 109/л, СОЭ — 18 мм/час.
Формула крови: базофилы — 0 %; эозинофилы — 0,5 %; палочкоядерные нейтрофилы — 4,5 %, сегментоядерные нейтрофилы — 44,5 %; лимфоциты — 38,5 %; моноциты— 11 %, плазматические клетки — 0,5 %, атипические мононуклеары — 0,5 %, ТЗН — 40 %.
Иммунограмма (27.07.10). Е-РОК. T-cells 34 % (N=40–60 %), или 1,15 ´ 109/л (N = 0,5–1), T-helpers 27 % (N =25–45%), или 0,91 ´ 109/л (N = 0,4–0,9). ЕАС-РОК. B-cells 32% (N = 15–30 %), или 1,08 ´ 109/л (N = 0,35–0,7), 0-cells 34 % (N = 10–20 %), или 1,14 ´ 109/л (N = 0,15–0,3).
Иммуноглобулины сыворотки крови (простая радиальная иммунодиффузия по Манчини): IgМ = 1,0г/л (N=0,8–1,6г/л); IgG = 10,0 г/л (N = 6,0–15,0г/л); IgA=2,2г/л (N=0,6–2,5 г/л); IgE = =30,1MЕ/мл (N=30–100 МЕ/мл).
Большие гранулярные лимфоциты— 2,25 %, или 0,198 ´ 109/л (N=0,25–0,40).
Показатель фагоцитоза нейтрофилов (латекс-тест) — 54 % (N = 60–80 %), фагоцитарное число нейтрофилов — 4,5у.е. (N = 5–10), количество активных фагоцитов — 1,8 ´ 109/л (N = 1,6–5,0 ´109/л), фагоцитарная емкость крови — 17,5 у.е. (N = 12,5–25,0).
HCT-тест: спонтанный — 279,5 (N=255,5 ± 8,2 у.е.), индуцированный (зимозан) — 41,4 % (N = 60–70 %).
Активность миелопероксидазы нейтрофилов — 9,92 (N= 18–23 у.е.), т.е. на 50 % меньше нижней границы нормы.
С-рективный белок (19.03.10) — 12 г/л (N — до 6 г/л), антистрептолизин О — 400 МЕ/мл (N — до 200 МЕ/мл).
Бак. посев (мазок с задней стенки глотки): b-гемоли-тический стрептококк — 104, кандида альбиканс— 105.
МРТ головного мозга (07.06.10): атрофия правого гиппокампа, венозная гемангиома правой теменной области.
Учитывая наличие лейкоцитоза, повышенной СОЭ, высокой концентрации С-реактивного белка и титра АСЛ-О, а также жалобы на упорные боли в суставах, мы пришли к выводу, что у пациентки помимо нейродегенеративного осложнения имел место синдром, сходный с ревматизмом, ассоциированный с персистенцией b-гемолитического стрептококка в ротоглотке, которая, в свою очередь, была опосредована имеющейся фагоцитарной недостаточностью.
Мать пациентки страдала ревматоидным артритом. Старший сын болеет рецидивирущим бронхитом, перенес затяжную прикорневую пневмонию, отмечаются рецидивирующий гайморит, ангины, атопический дерматит, медикаментозная аллергия.
HCT-тест (30.08.10): спонтанный — 243,5 (N=255,5±± 8,2у.е.), индуцированный (зимозан) — 56,8 % (N = 60–70%).
Активность миелопероксидазы нейтрофилов — 12,09 (N = 18–23 у.е.), т.е. на 30 % меньше нижней границы нормы.
Младший сын страдает хроническим ринофарингитом, тонзиллитом, рецидивирующими гайморитами, часто рецидивирующим herpes labialis (более 10 раз в году), атопическим дерматитом. Склонен к частым ОРВИ.
HCT-тест (30.08.10): спонтанный — 238,0 (N=255,5±± 8,2у.е.), индуцированный (зимозан) — 61,4 % (N = 60–70 %).
Активность миелопероксидазы нейтрофилов — 14,38 (N = 18–23 у.е.), т.е. на 25 % меньше нижней границы нормы.
Эти данные указывают на семейный характер недостаточности миелопероксидазы.
Больная Г.И.Б., 44 лет, первые симптомы заболевания отметила в 42-летнем возрасте с дрожания в правой руке, сначала возникавшем только при эмоциональном напряжении. Год спустя появилась скованность в левой ноге, нарушилась походка. Противопаркинсоническое лечение (проноран, неомидантан, сеган) не оказало должного эффекта. На момент осмотра: правосторонний гемипаркинсонизм, торсионная дистония в левой половине тела. В анамнезе: повторные выкидыши, периодически появляющаяся сыпь на поверхности всего тела неясного происхождения на протяжении последних 10 лет. Заболевания экстрапирамидной системы в семье отрицает.
Болеет хроническим ринофарингитом, тонзиллитом с регионарной лимфаденопатией, рецидивирующими ангинами, синуитами; отмечаются периоды субфебрилитета неясной этиологии, хронический аднексит, молочница.
Общий анализ крови (14.09.10): эр. — 4,3 ´ 1012/л; Нb— 137 г/л; ЦП — 0,96; тромбоциты — 237 ´ 109/л; лейкоциты— 3,7 ´ 109/л, СОЭ — 15 мм/час.
Формула крови: базофилы — 0 %; эозинофилы — 3,5%; палочкоядерные нейтрофилы — 5 %, сегментоядерные нейтрофилы — 49,0 %; лимфоциты — 38,0 %; моноциты— 3,5 %, плазматические клетки — 0,5 %, атипические мононуклеары — 0,5 %, ТЗН — 65 %.
Иммунограмма (14.09.10). Е-РОК. T-cells 26 % (N=40–60 %), или 0,36 ´ 109/л (N = 0,5–1), T-helpers 22 % (N =25–45 %), или 0,30 ´ 109/л (N = 0,4–0,9). ЕАС-РОК. B-cells 12% (N = 15–30 %), или 0,16 ´ 109/л (N = 0,35–0,7), 0-cells 62 % (N = 10–20 %), или 0,86 ´ 109/л (N = 0,15–0,3).
Иммуноглобулины сыворотки крови (простая радиальная иммунодиффузия по Манчини): IgМ = 1,6г/л (N =0,8–1,6 г/л); IgG = 13,5 г/л (N = 6,0–15,0 г/л); IgA=1,5г/л (N = 0,6–2,5 г/д); IgE = 28,5 MЕ/мл (N=30–100МЕ/мл).
Большие гранулярные лимфоциты — 5,75 %, или 0,213 ´ 109/л (N = 0,25–0,40).
Показатель фагоцитоза нейтрофилов (латекс-тест)— 70% (N = 60–80 %), фагоцитарное число нейтрофилов— 6,2 у.е. (N = 5–10), количество активных фагоцитов — 1,3´109/л (N = 1,6–5,0 ´ 109/л), фагоцитарная емкость крови— 11,2 у.е. (N = 12,5–25,0).
HCT-тест: спонтанный — 262,0 (N = 255,5 ± 8,2 у.е.), индуцированный (зимозан) — 51,7 % (N = 60–70 %).
Активность миелопероксидазы нейтрофилов — 12,5 (N= 18–23 у.е.), т.е. на 30 % меньше нижней границы нормы.
ПЦР сыворотки крови (15.09.10): HSV-1 положит., EBV положит., HHV-6 отрицат., HHV-7 положит., VZV отрицат.
МРТ головного мозга (06.01.10): фокальный энцефалитический очаг в правой лобной доли; 17.08.10: признаки воспаления хориоидального сплетения бокового желудочка.
Эти данные интерпретированы нами таким образом, что паркинсонизм был связан с дефицитом миелопероксидазы, а дистония — с реактивированной герпесвирусной нейроинфекцией, опосредованной имеющимся иммунодефицитом. Подтверждением герпесвирусной микст-нейроинфекции являются положительные результаты ПЦР сыворотки крови с видоспецифическими праймерами HSV-1, EBV и HHV-7, а также характерные изменения на нейровизуализационных снимках.
Больная О.В.Г., 42 лет, в 36-летнем возрасте после перенесенного стресса отметила скованность в правой руке. Наблюдалась гипомимия, акинезия исключительно в правой руке с преобладанием в кисти, мышечная ригидность в правых конечностях, больше — в руке, периодически появлялся тремор в правой кисти. Походка не была изменена. Нарушение обоняния больная отрицала. Отмечался положительный эффект на малые дозы агонистов дофаминовых рецепторов, которые больная самостоятельно отменила. В 40-летнем возрасте отметила изменение походки за счет скованности в правой ноге, однако старалась компенсировать болезненные симптомы занятиями гимнастикой. Повторно обратилась в 42-летнем возрасте, когда отмечались выраженные симптомы болезни Паркинсона с наличием выраженной гипокинезии и ригидностью, преимущественно в правых конечностях, тремором в правой руке и ноге, который при волнении генерализуется на все тело. Походка замедленна, нарушена за счет выраженной скованности в правой ноге. Постуральная неустойчивость — 2,5 по Хен и Яру. Положительный эффект на агонисты дофаминовых рецепторов. Болезнь Паркинсона в семье отрицает.
Страдает хроническим ринофарингитом, тонзиллитом, регионарной лимфаденопатией, периодической лихорадкой неясного генеза. В детстве на 1-м году жизни перенесла тяжелую двустороннюю бронхопневмонию, позже — двусторонний гнойный отит с затяжным течением, серию ангин и бронхитов. Более 10 лет страдала хроническим пиелонефритом, не связанным с конкрементами в мочевыводящих путях. Имеет место хронический гастродуоденит с частыми обострениями. Отмечаются аллергические проявления на домашнюю пыль, пыльцу растений, ряд пищевых продуктов. Перенесла отек Квинке, получала неотложную помощь.
Общий анализ крови (19.10.10): эр. — 4,2 ´ 1012/л; Нb— 140 г/л; ЦП — 1,0; тромбоциты — 420 ´ 109/л; лейкоциты— 10,1 ´ 109/л, СОЭ — 8 мм/час.
Формула крови: базофилы — 0,5 %; эозинофилы — 3,5%; палочкоядерные нейтрофилы — 3,5 %, сегментоядерные нейтрофилы — 65,0 %; лимфоциты — 21,5 %; моноциты — 6 %, ТЗН — 25 %.
Иммунограмма (19.10.10). Е-РОК. T-cells 23 % (N=40–60 %), или 0,49 ´ 109/л (N = 0,5–1,0), T-helpers 18% (N=25–45 %), или 0,39 ´ 109/л (N = 0,4–0,9). ЕАС-РОК. B-cells 30% (N = 15–30 %), или 0,65 ´ 109/л (N =0,35–0,7), 0-cells 47 % (N = 10–20 %), или 1,01 ´ 109/л (N = 0,15–0,3).
Иммуноглобулины сыворотки крови (простая радиальная иммунодиффузия по Манчини): IgМ = 1,2г/л (N=0,8–1,6 г/л); IgG = 11,5 г/л (N = 6,0–15,0 г/л); IgA=2,6г/л (N =0,6–2,5 г/л); IgE = 255,9,5 MЕ/мл (N=30–100 МЕ/мл).
Большие гранулярные лимфоциты — 0,5 %, или 0,05´109/л (N = 0,25–0,40).
Показатель фагоцитоза нейтрофилов (латекс-тест)— 80% (N = 60–80 %), фагоцитарное число нейтрофилов— 7,8 у.е. (N = 5–10), количество активных фагоцитов — 5,2´109/л (N = 1,6–5,0 ´ 109/л), фагоцитарная емкость крови — 51,2 у.е. (N = 12,5–25,0).
HCT-тест: спонтанный — 297,0 (N = 255,5 ± 8,2 у.е.), индуцированный (зимозан) — 38,5 % (N = 60–70 %).
Активность миелопероксидазы нейтрофилов — 13,3 (N= 18–23 у.е.), т.е. на 27 % меньше нижней границы нормы.
Таким образом, нами описана новая форма акинетико-ригидного синдрома — паркинсонизм у пациентов с дефицитом миелопероксидазы фагоцитов. Этот синдром характеризуется ранним началом в возрасте 40–45 лет у женщин, семейный анамнез которых отрицателен по болезни Паркинсона, но положительный по дефициту миелопероксидазы или, по крайней мере, по симптомам, которые могут быть интерпретированы как проявления этого иммунодефицита. Обычно у одного или сразу нескольких родственников пациентки также отмечается дефицит указанного фермента, поэтому обследование членов всей семьи играет важную роль в постановке диагноза. Чаще всего проявления экстрапирамидной недостаточности поначалу развиваются по гемитипу, а в дальнейшем асимметрия симптомов сохраняется даже при развитии двусторонних поражений. Ригидность превалирует над тремором, что является характерным признаком многих форм вторичного паркинсонизма. Отмечается удовлетворительная чувствительность к специфическим противопаркинсоническим препаратам, что указывает на дофаминергическую недостаточность как непосредственную причину двигательных расстройств в этих случаях.
Данные о нарушенной упаковке и постсинтетическом процессинге миелопероксидазы как причине фагоцитарной недостаточности при этом иммунодефиците перекликаются с основными пунктами ольфакторной теории паркинсонизма. Кроме того, у пациентов с дефицитом миелопероксидазы зачастую отмечается хроническое воспаление в зоне иннервации обонятельных нервов, что потенциально может влиять на адекватность упаковки молекул a-синуклеина, т.е. выступать в роли так называемых факторов внешней среды, опосредующих аномальную конфигурацию белков, избыточное накопление которых отмечается при акинетико-ригидном синдроме.
Приведенные нами данные согласуются с сообщениями о развитии синдрома паркинсонизма у пациентов с другими формами первичных иммунодефицитов, в частности при синдроме Чедиака — Хигаси и Х-сцепленной агаммаглобулинемии. Действительно, при синдроме Чедиака — Хигаси отмечается вторичный дефицит миелопероксидазы, связанный с затруднением доступа субстратов к этому ферменту из-за нарушения процесса фаголизосомального слияния, а иммуноглобулины, глубокий дефицит которых наблюдается при болезни Брутона, наделены свойствами опсонинов, т.е. способны опосредовать функцию иммунного фагоцитоза. И синдром Чедиака — Хигаси, и болезнь Брутона очень редко встречаются в человеческой популяции и сопровождаются слишком тяжелыми клиническими проявлениями инфекционного синдрома, чтобы претендовать на роль причины парксинсонизма у большого контингента пациентов с акинетико-ригидным синдромом, развившемся в третьей части жизни. Так, синдром Чедиака — Хигаси встречается с частотой 1:2000 000 000 [58], а Х-сцепленная агаммаглобулинемия— 1:200 000 населения развитых стран [64]. При этом большинство пациентов, страдающих первой болезнью, не доживают до 10-летнего возраста из-за развития бактериального сепсиса или лимфопролиферативных опухолей, вызванных герпесвирусами [57], а средняя продолжительность жизни людей с болезнью Брутона составляет 25–30 лет даже на фоне рациональной заместительной иммуноглобулинотерапии, так как зачастую развивается тяжелая легочно-сердечная недостаточность из-за прогрессирующего пневмосклероза [64]. Кроме того, Х-сцепленная агаммаглобулинемия наблюдается почти исключительно у лиц мужского пола, поэтому не может рассматриваться как весомая причина нейродегенеративных осложнений у женщин. Напротив, дефицит миелопероксидазы достаточно распространен в современной популяции, в частности, в США этот первичный иммунодефицит встречается с частотой 1: 2000 [41], а в Японии — немного реже, приблизительно с частотой 1:10 000 жителей, что, по-видимому, связано с определенной закрытостью популяции этой страны и ограниченным кровосмешением с представителями других этносов [43]. R.С. Cramer с соавт. сообщают о повышенной частоте этого первичного иммунодефицита в северной Италии, однако причина этого феномена все еще остается неизвестной [15]. Данные относительно частоты встречаемости дефицита миелопероксидазы в Украине отсутствуют, однако наши наблюдения свидетельствуют о довольно широкой распространенности этого заболевания в украинской популяции. В то же время глубина иммунной недостаточности при дефиците миелопероксидазы обычно невелика, что позволяет почти всем больным без специфического лечения доживать до такого возраста, когда потенциально могут проявиться нейродегенеративные осложнения.
Действительно, как показывают наши наблюдения, в возрасте 40–45 лет у некоторых таких пациентов дебютируют нейродегенеративные осложнения, в частности проявления паркинсонизма. Эти осложнения можно объяснить хорошо известной теорией нарушенной аутофагии, которая предложена для описания механизма нейродегенерации при болезнях Паркинсона, Альцгеймера и боковом амиотрофическом склерозе. Не следует забывать, что микроглия является производной моноцитов крови, поэтому имеющийся дефицит миелопероксидазы затрагивает и эти клетки [23]. Как сообщают Y.S. Kim и T.H. Joh (2006), микроглия на сегодняшний день рассматривается как ключевой тип клеток, ответственный за развитие нейродегенеративных процессов, так как именно они являются источником провоспалительных цитокинов, активных форм кислорода и эйкозаноидов, имеющих важное значение как в запуске, так и поддержании дистрофического процесса в нервной ткани [30]. Микроглия может также оказывать нейропротекторное воздействие, поэтому роль этих клеток при развитии нейродегенеративных осложнений трудно переоценить.
Существует экспериментальная модель, полученная на лабораторных животных, свидетельствующая о потенциальной возможности развития симптомов паркинсонизма при избирательном воздействии на миелопероксидазу глиальных клеток. Хотя большинство публикаций посвящено изучению активации миелопероксидазы, так как этот механизм рассматривают в качестве важного компонента патогенеза атеросклероза, гломерулонефрита, васкулитов и некоторых других тяжелых заболеваний, мы акцентируем внимание на клинических последствиях дефицита этого энзима, которые в некоторых случаях могут оказаться куда более опасными для здоровья человека, нежели эффекты его активации.
Среди непосредственных причин нейродегенерации при дефиците миелопероксидазы следует рассматривать нарушенную аутофагию, т.е. неэффективную обработку и утилизацию эндогенных молекул, в частности a-синуклеина в ткани ЦНС [38, 49], если речь идет о паркинсонизме, а также оксирадикальный стресс, сопровождающийся аномально интенсивным образованием активных форм кислорода, или воздействие нейротропных инфекционных агентов, репродукция которых опосредована имеющимися нарушениями иммунитета. Действительно, дефицит миелопер-оксидазы приводит к компенсаторной активации другого микробицидного фермента фагоцитов — НАДФ-оксидазы, опосредующего аномальную гиперпродукцию ряда активных форм кислорода, способных оказывать повреждающее воздействие на нейроны [14]. Компенсаторная гиперактивация НАДФ-оксидазы при дефиците миелопероксидазы убедительно продемонстрирована на примере инфекции, вызванной Aspergillus fumigatus [60]. Однако при инфекции, вызванной Candida albicans, напротив, доказана облигатная роль миелопероксидазы как микробицидного агента [2]. В то же время фагоцитарная недостаточность при этом первичном иммунодефиците может способствовать персистенции вирусных агентов в ткани ЦНС, так как микроглия является важным продуцентом интерферонов, а антигенная презентация, выполняемая этими клетками, способствует запуску и поддержанию локального иммунного ответа против вируса. S.J. Klebanoff и R.W. Coombs сообщают, что миелопероксидаза оказывает выраженное вируцидное действие на вирус иммунодефицита человека 1-го типа (HIV-1) благодаря индукции выработки гипохлорит-аниона, способствующего разрушению макромолекул патогена [33]. Аналогичные данные получены J.С. Chochola и соавт., которые показали, что репродукция HIV-1 прекращается при добавлении в культуру инфицированных клеток миелопероксидазы в дозе от 1,4 до 14,3 mU/ml, а дополнительное введение антиоксидантного фермента каталазы отменяет вируцидный эффект миелопероксидазы [13]. Как известно, HIV-1, персистируя в клетках микроглии, приводит к развитию прогрессирующей деменции, сходной по клиническим проявлениям с таковой при болезни Альцгеймера, поэтому важным представляется вопрос: не способствует ли дефицит миелопероксидазы развитию нейродегенеративных осложнений у ВИЧ-инфицированных пациентов?
Симптомы паркинсонизма у обследованных нами женщин возникли раньше, нежели это отмечается при классической болезни Паркинсона, но намного позже, чем при так называемой ювенильной форме паркинсонизма, имеющей, по-видимому, генетически детерминированную природу. Отдельного рассмотрения требует вопрос, почему симптомы нейродегенерации развиваются только после 40 лет, хотя признаки иммунной недостаточности имеются с момента рождения человека. Развитие симптомов нейродегенерации в третьей части жизни можно объяснить различной динамикой параллельно развивающихся патологических процессов, вызванных одной причиной, в частности быстрой динамикой клинических проявлений фагоцитарной недостаточности, связанной с нарушенным обезвреживанием антигенов микроорганизмов экзогенного происхождения, и медленным развитием синдрома накопления, обусловленного дефектной аутофагией некоторых эндогенно синтезированных молекулярных структур. Кроме того, учитывая, что в старшем возрасте появляются первые признаки атеросклероза, нельзя исключить роль сосудистого фактора и связанной с ним церебральной ишемии в декомпенсации имеющейся фагоцитарной недостаточности микроглии у пациентов с первичным иммунодефицитом, принимая во внимание тот факт, что миелопероксидаза использует именно молекулы кислорода для продукции микробицидных агентов. Более того, в третьей части жизни развивается так называемый возрастной иммунодефицит, сущность которого состоит в снижении активности клеточного звена адаптивного иммунитета из-за инволюции тимуса, что может отрицательно сказаться на первично скомпрометированной фагоцитарной функции микроглии, например, за счет снижения продукции гамма-интерферона Т-хелперами 1-го типа.
Также важным является вопрос, насколько глубина фагоцитарной недостаточности коррелирует с риском развития паркинсонизма в будущем. Логично было бы предположить, что чем тяжелее иммунная дисфункция, тем выше риск развития нейродегенеративных осложнений, однако здесь могут быть неожиданные результаты, например, в связи с воздействием ряда вмешивающихся факторов, в частности высокого риска летальности в раннем детском возрасте при глубокой иммунной недостаточности.
Возникновение синдрома паркинсонизма только у лиц женского пола с дефицитом миелопероксидазы, зафиксированное в данном сообщении, может быть исключительно случайным совпадением, связанным со спецификой набора пациентов, однако следует также учитывать, что эстрогены способны угнетать фагоцитарную активность, поэтому женский пол может быть отягощающим фактором, опо-средующим декомпенсацию имеющегося иммунодефицита.
Наше сообщение имеет не только теоретическое и диагностическое значение, но и позволяет по-новому взглянуть на способы терапии экстрапирамидных расстройств. Дефицит миелопероксидазы фагоцитов может быть купирован при длительной терапии препаратами рекомбинантного гамма-интерферона, модуляторов фагоцитоза по типу производных этиленпиперазина или гидрофталазина, а также иммуноглобулинов, учитывая опсонизирующие свойства антител. Важно установить, будет ли эффективной базисная терапия иммунодефицита по отношению к акинетико-ригидному синдрому, т.е. позволит ли компенсация фагоцитарной недостаточности достичь регресса неврологических симптомов, добиться остановки прогрессирования болезни или, по крайней мере, снизить темп углубления экстрапирамидной недостаточности. Еще более важным вопросом является установление факта, может ли раннее назначение базисной терапии иммунодефицита, т.е. начало лечения еще в детском возрасте, когда имеется лишь инфекционный синдром, предотвратить развитие тяжелых нейродегенеративных осложнений в будущем. Если будет получен положительный ответ хотя бы на один из этих вопросов, то мы будет иметь еще один прекрасный пример успешного применения последних достижений клинической нейроиммунологии для решения актуальных неврологических проблем.
1. Adler C.H., Stern M.B., Brooks M.L. Parkinsonism secondary to bilateral striatal fungal abscesses // Mov. Disord. — 1989. — Vol.4(4). — P. 333-337.
2. Aratani Y., Kura F., Watanabe H. et al. Critical role of myeloperoxidase and nicotinamide adenine dinucleotide phosphate-oxidase in high-burden systemic infection of mice with Candida albicans // J.Infect. Dis. — 2002. — Vol. 185(12). — P. 1833-1837.
3. Ball M.J. Limbic predilection in Alzheimer dementia: is reactivated herpesvirus involved? // Can. J. Neurol. Sci. Rep. — 1982.— Vol. 9. — P. 303-306.
4. Banks W.A. Immunotherapy and neuroimmunology in Alzheimer’s disease: a perspective from the blood-brain barrier // Immunotherapy. — 2010. — Vol. 2(1). — P. 1-3.
5. Bartels A.L., Leenders K.L. Neuroinflammation in the pathophysiology of Parkinson’s disease: evidence from animal models to human in vivo studies with [11C]-PK11195 PET // Mov. Disord.— 2007. — Vol. 22(13). — P. 1852-1856.
6. Braak H., Tredici K., Rub U. et al. Staging of brain pathology related to sporadic Parkinson’s disease // Neurobiol. Aging. — 2003.— Vol. 24. — P. 197-211.
7. Brookes J.T., Kanis A.B., Tan L.Y. et al. Cochlear implantation in deafness-dystonia-optic neuronopathy (DDON) syndrome // Int. J. Pediatr. Otorhinolaryngol. — 2008. — Vol. 72(1). — P. 121-126.
8. Burgos J.S., Ramirez C., Sastre I., Valdivieso F. Effect of apolipoprotein E on cerebral load of latent herpes simplex virus type 1 DNA// J. Virol. — 2006. — Vol. 80. — P. 5383-5387.
9. Carrazana E.J., Rossitch E. Jr, Samuels M.A. Parkinsonian symptoms in a patient with AIDS and cerebral toxoplasmosis// J. Neurol. Neurosurg. Psychiatry. — 1989. — Vol. 52(12). — P. 1445-1447.
10. Casrouge А., Zhang S.Y., Eidenschenk С. et аl. Herpes simplex virus encephalitis in human UNC-93B deficiency // Science. — 2006.— Vol. 314. — P. 308-312.
11. Cech P., Stalder H.S., Widmann J.J., Rohrer A., MiescherP.A. Leukocyte myeloperoxidase deficiency and diabetes mellitus associated with Candida albicans liver abscess // Am. J. Med. — 1979.— Vol.66. — P. 149-153.
12. Chen H., Jacobs E., Schwarzschild M.A. Nonsteroidal antiinflammatory drug use and the risk for Parkinson's disease // Ann. Neurol. — 2005. — Vol. 58(6). — P. 963-967.
13. Chochola J., Yamaguchi Y., Moguilevsky N. Virucidal effect of myeloperoxidase on human immunodeficiency virus type 1-infected T cells // Antimicrob. Agents. Chemother. — 1994. — Vol. 38(5). — P.969-972.
14. Clark S.R., Coffey M.J., Maclean R.M. et al. Characterization of nitric oxide consumption pathways by normal, chronic granulomatous disease and myeloperoxidase-deficient human neutrophils // J. Immunol. — 2002. — Vol. 169(10). — P. 5889-5896.
15. Cramer R., Soranzo M.R., Dri P. et al. Incidence of myeloperoxidase deficiency in an area of northern Italy: histochemical, biochemical and functional studies // Brit. J. Haemat. — 1982. — Vol.51. — P. 81-87.
16. Danik M., Champagne D., Petit-Turcotte C. et al. Brain lipoprotein metabolism and its relation to neurodegenerative disease // Crit. Rev. Neurobiol. — 1999. — Vol. 13. — P. 357-407.
17. Davis L.E., Johnson R.T. An explanation for the localization of herpes simplex encephalitis? // Ann. Neurol. — 1979. — Vol.5.— P. 2-5.
18. Donati D., Akhiani N., Fogdell-Hahn A. et al. Detection of human herpesvirus-6 in mesial temporal lobe epilepsy surgical resections// Neurology. — 2003. — Vol. 61(10). — P. 1405-1411.
19. Fotheringham J., Donati D., Akhyani N. et al. Association of human herpesvirus-6-B with mesial temporal lobe epilepsy // PLoS Med. — 2007. — Vol. 4(5). — e180.
20. Fotheringham J., Williams E.L., Akhyani N., Jacobson S. Human herpesvirus 6 (HHV-6) induces dysregulation of glutamate uptake and transporter expression in astrocytes // J. Neuroimmune Pharmacol. — 2008. — Vol. 3(2). — P. 105-116.
21. Ganesalingam J., Lacomis D., Lustgarten J. et al. PATH45 Cytoskeletal and inflammatory protein biomarkers for amyotrophic lateral sclerosis // J. Neurol. Neurosurg. Psychiatry. — 2010. — Vol.81(11). — e20.
22. Hamers M.N., Bot A.A., Weening R.S. et al. Kinetics and mechanism of the bactericidal action of human neutrophils against Escherichia coli // Blood. — 1984. — Vol. 64(3). — P. 635-641.
23. Hanisch U.K., Kettenman H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain // Nature Neurosci. — 2007. — Vol. 10(11). — P. 1387-1394.
24. Hauser R.A., Friedlander J., Baker M.J. et al. Adult Chediak-Higashi parkinsonian syndrome with dystonia // Mov. Disord. — 2000.— Vol. 15(4). — P. 705-708.
25. Itzhaki R.F., Cosby S.L., Wozniak M.A. Herpes simplex virus type 1 and Alzheimer’s disease: the autophagy connection // J. Neurovirol. — 2008. — Vol. 14(1). — P. 1-4.
26. Itzhaki R.F., Lin W.R., Sang D., Wilcock G.K. et al. Herpes simplex virus type 1 in brain and risk of Alzheimer’s disease // Lancet.— 1997. — Vol. 349. — P. 241-244.
27. Jacobi C., Koerner C., Fruehauf S. et al. Presynaptic dopaminergic pathology in Chediak-Higashi syndrome with parkinsonian syndrome // Neurology. — 2005. — Vol. 64(10). — P. 1814-1815.
28. Kameoka Y., Persad A.S., Suzuki K. Genomic variations in myeloperoxidase gene in the Japanese population // Jpn. J. Infect. Dis. — 2004. — Vol. 57(5). — S12-13.
29. Karatas H., Gurer G., Pinar A. et al. Investigation of HSV-1, HSV-2, CMV, HHV-6 and HHV-8 DNA by real-time PCR in surgical resection material of epilepsy patients with mesial temporal lobe sclerosis// J. Neurol. Sci. — 2008. — Vol. 264(1–2). — P. 151-156.
30. Kim Y.S., Joh T.H. Microglia, major player in the brain inflammation: their roles in the pathogenesis of Parkinson’s disease // Exp. Mol. Med. — 2006. — Vol. 38(4). — P. 333-347.
31. Kitahara M., Eyre H.J., Simonian Y., Atkin C.L., HasstedtS. Hereditary myeloperoxidase deficiency // Blood. — 1981. — Vol.57.— P. 888-893.
32. Klebanoff S.J. Myeloperoxidase // Proc. Assoc. Am. Physicians. — 1999. — Vol. 111(5). — P. 383-389.
33. Klebanoff S.J., Coombs R.W. Viricidal effect of polymorphonuclear leukocytes on human immunodeficiency virus-1. Role of the myeloperoxidase system // J. Clin. Invest. — 1992. — Vol. 89(6). — P.2014-2017.
34. Koenigsknecht-Talboo J., Landreth G.E. Microglial phagocitosis induced by fibrillar b-amyloid and IgGs are differentially regulated by proinflammatory cytokines // J. Neurosci. — 2005. — Vol.25. — P.8240-8249.
35. Lerner A., Bagic A. Olfactory pathogenesis of idiopathic Parkinson disease revisited // Mov. Disod. — 2008. — Vol. 8. — P.1076-1084.
36. Maggi P., de Mari M., Moramarco A. et al. Parkinsonism in a patient with AIDS and cerebral opportunistic granulomatous lesions// Neurol. Sci. — 2000. — Vol. 21(3). — P. 173-176.
37. Mamishi S., Shahmahmoudi S., Tabatabaie H. Novel BTK mutation presenting with vaccine-associated paralytic poliomyelitis // Eur. J. Pediatr. — 2008. — Vol. 167(11). — P. 1335-1338.
38. Mariсo G., Madeo F., Kroemer G. Autophagy for tissue homeostasis and neuroprotection // Curr. Opin. Cell. Biol. — 2010 [Epub ahead of print].
39. Matsuura K., Kabuto H., Makino H., Ogawa N. Initial cyclosporin A but not glucocorticoid treatment promotes recovery of striatal dopamine concentration in 6-hydroxydopamine lesioned mice// Neurosci. Lett. — 1997. — Vol. 230(3). — P. 191-194.
40. Matsuura K., Makino H., Ogawa N. Cyclosporin A attenuates the decrease in tyrosine hydroxylase immunoreactivity in nigrostriatal dopaminergic neurons and in striatal dopamine content in rats with intrastriatal injection of 6-hydroxydopamine // Exp. Neurol. — 1997.— Vol. 146(2). — P. 526-535.
41. Nauseef W.M. Myeloperoxidase deficiency // Hemat. Oncol. Clin. North Am. — 1988. — Vol. 2. — P. 135-158.
42. Nauseef W.M., Brigham S., Cogley M. Hereditary myeloperoxidase deficiency due to a missense mutation of arginine 569 to tryptophan // J. Biol. Chem. — 1994. — Vol. 269. — P. 1212-1216.
43. Nunoi H., Kohi F., Kajiwara H. et al. Prevalence of inhe-rited myeloperoxidase deficiency in Japan // Microbiol. Immunol.— 2003.— 47(7). — P. 527-531.
44. Ohashi Y.Y., Kameoka Y., Persad A.S. et al. Novel missense mutation found in a Japanese patient with myeloperoxidase deficiency// Gene. — 2004. — Vol. 327(2). — P. 195-200.
45. Ohno H. Association of primary myeloperoxidase deficiency and myeloproliferative neoplasm // Intern. Med. — 2010. — Vol.49(22).— P. 2527-2528.
46. Ostanin D.V., Barlow S., Shukla D., Grisham M.B. NADPH-oxidase but not myeloperoxidase protects lymphopenic mice from spontaneous infections // Biochem. Biophys. Res. Commun. — 2007.— Vol.355(3). — P. 801-806.
47. Papapetropoulos S., Friedman J., Blackstone C. et al. Aprogressive, fatal dystonia-Parkinsonism syndrome in a patient with primary immunodeficiency receiving chronic IVIG therapy // Mov. Disord. — 2007. — Vol. 22(11). — P. 1664-1666.
48. Persad A.S., Kameoka Y., Kanda S. Arginine to cysteine mutation (R499C) found in a Japanese patient with complete myeloperoxidase deficiency // Gene. Expr. — 2006. — Vol. 13(2). — P. 67-71.
49. Plowey E.D., Chu C.T. Synaptic dysfunction in genetic models of Parkinson’s disease: A role for autophagy? // Neurobiol. Dis. — 2010 [Epub ahead of print].
50. Prusiner S.B. Shattuck lecture — neurodegenerative disease and prions // N. Engl. J. Med. — 2001. — Vol. 344. — P. 1516-1526.
51. Quartier P., Foray S., Casanova J.L. Enteroviral meningoencephalitis in X-linked agammaglobulinemia: intensive immunoglobulin therapy and sequential viral detection in cerebrospinal fluid by polymerase chain reaction // Pediatr. Infect. Dis. J. — 2000.— Vol.19(11).— P. 1106-1108.
52. Quinn N. Pseudoparkinsonian tremor and dysgammaglobulinaemic polyneuropathy // J. R. Soc. Med. — 1990. — Vol. 83(10).— P.673-674.
53. Rosen H., Michel B.R. Redundant contribution of myeloper-oxidase-dependent systems to neutrophil-mediated killing of Escherichia coli // Infect. Immun. — 1997. — Vol. 65(10). — P. 4173-4178.
54. Silveira-Moriyama L., Moriyama T.S., Gabbi T.V. et al. Chediak-Higashi syndrome with parkinsonism // Mov. Disord. — 2004. — Vol. 19(4). — P. 472-475.
55. Theodore W.H., Epstein L., Gaillard W.D. et al. Human herpes virus 6B: a possible role in epilepsy? // Epilepsy. — 2008. — Vol.49(11). — P. 1828-1837.
56. Uesugi H., Shimizu H., Maehara T., Arai N., Nakayama H. Presence of human herpesvirus 6 and herpes simplex virus detected by polymerase chain reaction in surgical tissue from temporal lobe epileptic patients // Psychiatry Clin. Neurosci. — 2000. — Vol.54(5).— P.589-593.
57. Uyama E., Hirano T., Ito K. et al. Adult Chйdiak-Higashi syndrome presenting as parkinsonism and dementia // Acta Neurol. Scand. — 1994. — Vol. 89(3). — P. 175-183.
58. Uyama E., Hirano T., Yoshida A. et al. An adult case of Chйdiak-Higashi syndrome with parkinsonism and marked atrophy of the central nervous system // Rinsho Shinkeigaku. — 1991. — Vol.31(1). — P. 24-31.
59. Vezzani A., Granata T. Brain inflammation in epilepsy; experimental and clinical evidence // Epilepsia. — 2005. — Vol.46(11).— P.1724-1743.
60. Washburn R.G., Gallin J.I., Bennett J.E. Oxidative killing of Aspergillus fumigatus proceeds by parallel myeloperoxidase-dependent and -independent pathways // Infect. Immun. — 1987.— Vol.55(9).— P. 2088-2092.
61. Weil S.C., Rosner G.L., Reid M.S. et al. Translocation and rearrangement of myeloperoxidase gene in acute promyelocytic leukemia // Science. — 1988. — Vol. 240. — P. 790-792.
62. Wozniak M.A., Mee A.P., Itzhaki R.F. Herpes simplex virus type 1 DNA is located within Alzheimer’s disease amyloid plaques // J. Pathol. — 2009. — Vol. 217(1). — P. 131-138.
63. Zhang S.Y., Jouanguy E., Ugolini S. et al. TLR3 deficiency in otherwise healthy patients with herpes simplex encephalitis // Science.— 2007. — Vol. 317. — P. 1522-1527.
64. Ziegner U.H., Kobayashi R.H., Cunningham-Rundles C. Progre