
Международный неврологический журнал 1 (39) 2011
Вернуться к номеру
Клиническое наблюдение поздней диагностики болезни Реклингхаузена у 18-летнего юноши
Авторы: Шнайдер Н.А., д.м.н., проф.; Шаповалова Е.А., ГОУ ВПО «Красноярский государственный медицинский университет им. проф. В.Ф. Войно-Ясенецкого Минздравсоцразвития РФ», кафедра медицинской генетики и клинической нейрофизиологии ИПО, Неврологический центр эпилептологии, нейрогенетики и исследования мозга, г. Красноярск, Молгачев А.А., Лечебно-диагностический центр Международного института биологических систем, г. Красноярск, Шаповалова Л.П., Федеральное государственное учреждение «Главное бюро по Красноярскому краю», филиал № 35
Рубрики: Неврология
Версия для печати
Статья посвящена актуальной проблеме детской неврологии — наследственному нейрокожному синдрому — нейрофиброматозу 1-го типа (болезни Реклингхаузена). Кратко освещены дефиниция, эпидемиология, этиопатогенез и клинические проявления заболевания. Приведен клинический разбор случая поздней диагностики заболевания у 18-летнего юноши. Подчеркнута важность междисциплинарного подхода к диагностике и диспансерному наблюдению детей с нейрофиброматозом 1-го типа и членов их семей — носителей мутантного гена.
Нейрокожный синдром (факоматоз), нейрофиброматоз 1-го типа (болезнь Реклингхаузена), NF1, педиатрия, клинический случай.
Введение
Факоматозы (греч. phakos — пятно) — наследственные болезни, объединенные общими звеньями патогенеза. Первые описания факоматозов встречаются в середине XIX века (1862 г.). В настоящее время описано около 60 нозологических самостоятельных форм [1–3]. По этиопатогенезу факоматозы делятся на ангиоматозы и бластоматозы. Нейрофиброматоз относится к бластоматозам и представляет собой наиболее распространенную форму моногенной наследственной патологии, встречается в популяции с частотой от 1 на 2000 до 1 на 4000. Тип наследования заболевания аутосомно-доминантный с пенетрантностью, близкой к 100 % [4–7].
Нейрофиброматоз 1-го типа (син.: классический нейрофиброматоз, периферический нейрофиброматоз, онтогенная дистрофия, нейрофибролипоматоз, глиофиброматоз, нейроглиоматоз, врожденная нейроэктодермальная дисплазия Ван-Богарта, болезнь Ватсона, синдром нейрофиброматоза — феохромоцитомы — дуоденального карциноида, NF1) впервые описан немецким врачом Frederich von Recklinghausen в 1882 году. В настоящее время заболевание известно как болезнь Реклингхаузена (англ. Von Recklinghausen’s disease, Von Recklinghausen neurofibromatosis) [8]. Частота встречаемости NF1 — 1 на 3000 населения.
Этиопатогенез
NF1 — аутосомно-доминантное заболевание с высокой пенетрантностью и высокой частотой возникновения новых мутаций (OMIM 162200) [9]. Примерно 50% случаев заболевания представляют собой мутации denovo. Высокая частота спонтанных мутаций объясняется большими размерами гена и/или определенными особенностями его внутренней структуры. В 1990–1995гг. была локализована мутация на хромосоме 17q11.2. Ген NF1 является достаточно протяженным и сложно организованным. Он имеет длину около 350кб, состоит из 60 экзонов и экспрессируется, помимо нервной системы, в различных тканях. Ген кодирует белок нейрофибромин, являющийся супрессором опухолевого роста [10]. Нейрофибромин продуцируется в нервных клетках и специализированных клетках нейроглии (олигодендроцитах и шванновских клетках). Белок содержит в своем составе домен белков-активаторов ГТФазы. Посредством этого домена нейрофибромин в норме взаимодействует с продуктом проонкогена RAS, ингибируя его функцию и реализуя свой супрессорный эффект в отношении клеточной пролиферации [11–15]. У больных NF1 описано свыше 500 различных мутаций в гене на хромосоме 17q. Эти мутации нарушают регулирующую роль гена NF1 в каскаде событий онкогенеза. Характер мутаций весьма специфичен: более 80 % из них ведут к синтезу нефункционального «усеченного» белка либо к полному отсутствию транскрипта (нонсенс-мутации, мутации в сайтах сплайсинга, делеции и вставки со сдвигом рамки, крупные делеции, охватывающие весь ген или его значительную часть). Остальные мутации представляют собой внутренние делеции без сдвига рамки и миссенс-мутации, затрагивающие функционально важные участки нейрофибромина. Мутации распределены в пределах кодирующей области NF1 достаточно равномерно, и лишь экзоны 10а-10с.31 и 37 представляют собой области с относительно высокой частотой повреждения (от 6 до 30 % всех выявленных мутаций). Описаны также случаи NF1, обусловленного цитогенетическими перестройками, затрагивающими критический хромосомный сегмент 17q11.2 [16, 17].
Клинические проявления заболевания
Типичные для NF1 плоские пигментные пятна носят характер пятен цвета кофе с молоком (франц. cafe-au-lait; англ. milk coffee) и «веснушчатых гроздьев» на коже, вариабельных размеров, окраски, расположены на различных участках тела, с четкими границами, как правило, неправильной формы, обычно появляются к двухлетнему возрасту. Узелки Лиша (англ. Lisch nodules) патогномоничны пигментным пятнам на радужке глаза (гамартомы), выявляются при офтальмологическом осмотре с помощью щелевой лампы. Узелки Лиша чаще выявляются в более зрелом возрасте, например в возрасте от 0 до 4 лет — до 22 % случаев; 5–9 лет — до 41 %; 10–19 лет — до 85 %; старше 20 лет — до 95 % больных NF1. Как правило, пятна цвета кофе с молоком и узелки Лиша безопасны для здоровья человека. Иногда пигментные кожные пятна являются единственным проявлением NF1, так как небольшие нейрофибромы не всегда удается обнаружить, особенно в детском возрасте. Считается, что наличие не менее 6 пигментных пятен диаметром не менее 1,5 см позволяет диагностировать NF1 при отсутствии каких-либо других симптомов.
Одним из проявлений заболевания являются множественные нейрофибромы по ходу периферических нервов в виде болезненных округлых узелков в толще кожи, вариабельных по форме, размерам (от просяного зерна до 5 см и более) и локализации [17–20]. Выявляемость нейрофибром зависит от возраста больных: до 10 лет — 14 %, от 10 до 19 лет — 44 %, 20–29 лет — 85 %, старше 30 лет — 94 %. Первые нейрофибромы появляются в период препубертата или пубертата. С возрастом отмечается неуклонный медленный рост нейрофибром, особенно заметный в период полового созревания индивидуума, а также у женщин в период беременности. При пальпации нейрофибромы часто безболезненны, но если в патологический процесс вовлечены периферические нервы, то возникают боли, гипестезии. Опухоль смещается только в поперечном направлении вместе с нервным стволом. При этом возникают иррадиирующие боли в зоне иннервации. В некоторых случаях нейрофиброматоз может носить весьма ограниченный сегментарный характер (например, медиастинальные нейрофибромы в сочетании с опухолями в соответствующем кожном сегменте), но чаще он является генерализованным (опухоли на туловище, шее, голове, конечностях). Склонность к злокачественному перерождению отмечается у 3–15 % больных NF1 [21, 22]. Существуют плексиформные нейрофибромы, которые представляют собой разрастание тканей нерва в строме из нормальных окружающих тканей, сопровождаются гипертрофией пораженных участков тела (слоновостью) и внутренних органов, обычно одиночные, больших размеров. Частота встречаемости около 5%. Чаще всего они начинают развиваться до рождения ребенка и становятся очевидными к двухлетнему возрасту. Также к проявлениям NF1 относятся папилломы, но они встречаются редко [23, 24].
Несмотря на периферический характер NF1, у части больных может наблюдаться вовлечение центральной и периферической нервной системы с развитием опухолей— астроцитом и глиом зрительных путей, эпендимом, менингиом, нейролеммом, шванном, спинальных нейрофибром [23, 25, 26]. Оптическая глиома— доброкачественная опухоль зрительного нерва редко встречается у детей младшего возраста, чаще дебютирует в десятилетнем возрасте в виде постепенного снижения зрения. При NF1 могут выявляться опухоли иной локализации, включая феохромоцитому. Мутация гена NF1 может быть причиной миелодиспластического синдрома и редкого типа лейкоза— ювенильной миеломоноцитической лейкемии (англ. juvenile myelomonocytic leukemia — JMML), являющейся патогномоничной для детей моложе двух лет. Дети с JMML жалуются на быструю утомляемость, усталость, лихорадку, частые кровотечения (гематомы) при незначительных травмах [27]. Для NF1 характерны также дополнительные клинические проявления: в 50 % случаев — когнитивные нарушения различной степени (от легких до выраженных), чаще в сочетании с негрубым или умеренным снижением IQ, затруднением в освоении письма, чтения, математики [28]; эндокринные расстройства (феохромоцитома, нарушение роста и полового созревания); изменения скелета (сколиоз — до 15 %, деформация грудной клетки, спондилолистез, незаращение дужек позвонков, краниовертебральные аномалии, асимметрия черепа, псевдоартроз); эпилептические приступы и др. [29–31]. Могут быть частые переломы костей конечностей, которые тяжело поддаются лечению (долго срастаются) и требуют консультации ортопеда. У детей с NF1 может быть снижен мышечный тонус, негрубо нарушена координация движений. Характерен набор определенных стигм дизэмбриогенеза: большой размер черепа (окружность головы) — более 4 стандартных отклонений, чем обычно в данном возрасте, низкий рост, гипертелоризм, антимонголоидный разрез глаз, низко посаженные уши, шейный птеригиум, стеноз легочной артерии. Такие дети могут быть малоинициативными и менее эмоциональны по сравнению со здоровыми сверстниками [32, 33].
При постановке диагноза NF1 рекомендуется использовать диагностические критерии, рекомендованные Международным комитетом экспертов по нейрофиброматозу [34, 35]. Таким образом, NF1 может быть диагностирован при наличии у больного не менее 2 из следующих признаков: 1) не менее 5 пигментных пятен цвета кофе с молоком диаметром более 5 мм у детей препубертатного возраста и не менее 6 пятен диаметром более 1,5 см в постпубертатном возрасте; 2) две и более нейрофибромы любого типа или одна плексиформная нейрофиброма; 3) веснушчатость в подмышечных или паховых складках; 4) дисплазия крыла клиновидной кости или врожденное истончение кортикального слоя длинных костей с псевдоартрозом или без него; 5) глиома зрительного нерва; 6) два и более узелка Лиша на радужке при исследовании с помощью щелевой лампы; 7) наличие у родственников первой степени родства NF1 по тем же критериям.
Клинический случай. Больной П., 18 лет, обратился на прием в Неврологический центр эпилептологии, нейрогенетики и исследования мозга Университетской клиники КрасГМУ им. проф. В.Ф. Войно-Ясенецкого в январе 2010 г. с жалобами на постепенно прогрессирующее расстройство равновесия, в том числе при ходьбе, метеозависимые головные боли с кризовым течением (ликвородинамические кризы), а также с 2007 г. — приступообразные насильственные повороты головы и глаз влево на фоне сохранного сознания без падений и ретроградной амнезии, частота которых постепенно нарастала в течение последних 2 лет.
Из анамнеза известно, что с ранних детских лет у ребенка отмечались множественные пятна гиперпигментации цвета кофе с молоком: на переднебоковой поверхности живота слева крупное пятно (25´19см) с четкими неровными контурами, мелкие пятна вариабельных размеров на спине и конечностях (более 10), которые игнорировались при диспансерном наблюдении ребенка у участкового педиатра и дерматолога по месту жительства ребенка. Размеры пятен гиперпигментации участковым педиатром и другими лечащими врачами по месту дальнейшего наблюдения и лечения пациента не измерялись, их количество не уточнялось, что затрудняло оценку их роста в динамике по мере взросления пациента. Однако при тщательном сборе анамнеза на момент настоящей консультации, со слов отца пациента, отмечена тенденция к увеличению числа и размеров пятен гиперпигментации в течение последних 2–3 лет. Несмотря на типичную клиническую симптоматику дебюта заболевания с кожных проявлений в раннем детском возрасте, ребенок не был направлен участковым педиатром и дерматологом на консультацию к нейрогенетику, не проведена МРТ головного мозга для исключения бессимптомного (малосимптомного) поражения ЦНС на ранних стадиях развития патологического процесса.
Пробанд до 12-летнего возраста (до 2004 г.) считался здоровым ребенком, рос и развивался соответственно возрасту и полу. В 2004 г. у ребенка появились и стали постепенно прогрессировать расстройства равновесия, на фоне чего мальчик поскользнулся, упал и ударился затылочной областью об лед. В первые сутки после ЗЧМТ развилась общемозговая симптоматика в виде тошноты и головной боли, но за медицинской помощью родители не обращались, лечили ребенка у бабушки-знахарки — общемозговая симптоматика сохранялась, в связи с чем через неделю ребенок был госпитализирован в детское нейрохирургическое отделение ГБ №20 им. Берзона г. Красноярска, где впервые по данным нейровизуализации было диагностировано объемное образование червя мозжечка.
В связи со сложной локализацией объемного образования по желанию родителей ребенок был госпитализирован и прооперирован в Германии в Клиническом комплексе г. Нюрбернга: в июне 2004г. в условиях интубационного наркоза проведена резекция объемного образования червя мозжечка. Объемное образование удалено при использовании операционного микроскопа CUSA с сильным увеличением. Опухоль пересекала срединную линию справа на 1,5 см в области полушария, слева — на 2–2,5 см. Краниально опухоль достигала в области верхнего отдела червя мозжечка, поверхности мозга. Рострально опухоль достигла крыши четвертого желудочка с обеих сторон, от начальной части водопровода до половины крыши желудочка снизу, где она достигла основания четвертого желудочка. Интраоперационно проведен забор биопсийного материала для срочного гистологического исследования и для развернутой гистологии. Оперативное вмешательство выполнено без особенностей, послеоперационный период прошел без осложнений. По результатам срочного гистологического исследования сделано предположение о медуллобластоме IV степени. На основании чего в послеоперационном периоде, не дожидаясь результатов развернутого гистологического и иммуногистохимического исследований, ребенку начали проведение радиотерапии при использовании техники нейрооси (доза 9,6 Gy). Побочных явлений не отмечено, но во время проведения радиотерапии были получены результаты развернутого гистологического и иммуногистохимического исследования биопсийного материала опухоли и предлежащих тканей головного мозга из Университетской клиники г. Мюнстера — объемное образование описано как плотная нейроэктодермальная опухоль. Опухолевые клетки содержали преимущественно круглые, мономорфные ядра и прозрачную (чистую) цитоплазму. Повсюду визуализированы узкие периваскулярные ареалы, но без типичных периваскулярных псевдорозеток. Выявлены единичные митозы (менее 1 митоза на 10 клеток). Фокально отмечались псевдоцистические флокуляции (хлопья). В одном образце ткани распознано четкое астроцитарное дифференцирование с мультиполярными продолжениями и отдельными эозинофильными гранулированными или гомогенными телами; типичные волокна Розенталя не видны, как и бифазная структура. По краю — полный опухолевый некроз. Сквозные выраженные сосудистые пролифераты, частично с грамелуроидальными структурами и гирляндным нанизыванием. Выявлены ограниченные отложения гемосидерина как показатель кровотечения. Опухоль была ограничена реактивно измененной тканью мозжечка. По результатам иммуногистохимического анализа обнаружены прозрачные клеточные участки, являющиеся преимущественно, но не исключительно, GPAF-отрицательными, тогда как при рутинном окрашивании в отчетливо видимых астроцитарных ареалах были обнаружены значительные DPAF-положительные результаты. Клетки опухоли были слабопозитивны к нейроспецифической энолазе (NSE). Пролиферация KL67/M1B1 составила в среднем 3 %. На основании данных гистологического и иммуногистохимического анализа, локализации опухоли и возраста пациента экспертами Университетской клиники г. Мюнстера опухоль была оценена как пилоцетарная астроцитома (1-й степени). Необычным для этого диагноза было наличие доминирующих прозрачных компонентов (мукоидной дегенерации). Однако оснований для медуллобластомы не выявлено.
В связи с тем, что мнения экспертов при проведении срочного гистологического и развернутого гистологического и иммуногистохимического исследований разошлись, для обсуждения дальнейшей тактики лечения пациента был привлечен консультант (педиатр клиники г. Нюрбернга), после проведения консилиума было принято решение о приостановке радиотерапии, рекомендована консультация одного из ведущих специалистов-невропатологов Германии. Проведено повторное гистологическое исследование в Университетской клинике г. Бонна, по результатам которого иммунофенотип опухолевой ткани свидетельствовал против эпендимальной опухоли. Бифазное строение опухоли было наиболее типично для пилоцитарной астроцитомы с прорастанием в мозжечок. На основании локально повышенных показателей KL67-маркировки объемное образование было охарактеризовано как пилоцитарная астроцитома с повышенной активностью пролиферации (2-й степени). На основании вышеперечисленного дифференцированы пилоцитарная астроцитома и опухоль группы эпендимомы. Данных за десмопластическую медуллобластому не выявлено.
Так как по результатам контрольного гистологического и иммуногистохимического исследования разногласий у специалистов не возникло, был сформулирован заключительный клинический диагноз: пилоцетарная астроцитома 2-й степени. Дальнейший прогноз специалистами был расценен как благоприятный, так как опухоль задней черепной ямки (червя мозжечка) была удалена, а при астроцитоме 2-й степени нет необходимости в дальнейшем лечении и диспансерном наблюдении.
Таким образом, в 2004 г. клинический диагноз был сформулирован как изолированное поражение ЦНС, несмотря на наличие типичных кожных проявлений нейрофиброматоза 1-го типа — множественных крупных пятен гиперпигментации цвета кофе с молоком в сочетании с поражением ЦНС (нейроэпителиальной опухолью ствола головного мозга), не был проведен клинико-генеалогический анализ родословной пробанда, что повлекло за собой ошибочную тактику ведения больного, трактовку специалистами благоприятного прогноза и отказ от диспансерного наблюдения пациента у нейрогенетика, нейрохирурга, поскольку для данной патологии характерен высокий риск рецидивирования доброкачественных опухолей головного мозга (в данном клиническом случае — пилоцетарной астроцитомы червя мозжечка), включая образование новых опухолей центральной и периферической нервной системы, включая полушария большого мозга, зрительные нервы, спинной мозг, спинномозговые корешки и периферические нервы, а также доброкачественные опухоли другой локализации (нейрофибромы, шванномы и др.).
В связи с тем, что отцу пробанда было сообщено о «благоприятном прогнозе» заболевания, больной после возвращения в Россию в течение последующих 3 лет на диспансерном учете по месту жительства не состоял. Однако в 2005–2006 гг. заболевание неуклонно прогрессировало, о чем свидетельствовали (со слов отца пробанда) увеличение численности и размеров пятен гиперпигментации цвета кофе с молоком на коже больного и вновь появившаяся общемозговая и очаговая неврологическая симптоматика (рецидив астроцитомы). С 2005 г. пациенту проводились МРТ- и КТ-исследования головного мозга в динамике. МРТ головного мозга (0,5 Тс) от 21.11.2005г. (Красноярск): в области удаления опухоли ствола мозга обнаружен тканевой участок до 0,8см, не накапливающий контраст, рекомендована МРТ в динамике через 2 мес. МРТ головного мозга (0,5 Тс) от 27.03.2006 г. (Красноярск): отмечен продолженный рост опухоли в виде тканевого образования 1,5 см. МРТ головного мозга (0,5 Тс) с контрастированием через год, от 26.03.2007г. (Красноярск): выявлено более активное включение контрастного вещества (гадолиния) в области патологического образования мозжечка, при этом размеры образования были прежними — 1,5 см. В связи с этим проведена повторная МРТ с мощностью магнитного поля 1,5 Тс и с контрастированием гадолинием (омнискан в/в 10мл) от 13.09.2007 г. (Красноярск): в области постоперационного вмешательства определялась обширная зона послеоперационных кистозно-глиозно-атрофических изменений размером до 5,4 ´4,4´2,8 см, в области послеоперационных изменений в краниальных отделах червя мозжечка, больше слева, определялся неоднородный гиперинтенсивный сигнал по Т2 и изогипоинтенсивный по Т1, визуализировано объемное образование с нечеткими, неровными контурами размером 2,5´2,3´ 1,6 см с умеренно выраженной перифокальной реакцией, несколько компремирующее правую половину моста и правую ножку мозга. После внутривенного введения контраста определялся периферический тип усиления интенсивности сигнала в области ранее вышеописанного образования. Кроме того, впервые в медиальных отделах левой височной доли выявлено объемное образование неоднородно гиперинтенсивного сигнала по Т2 и неоднородного гипоинтенсивного по Т1, с нечеткими и неровными контурами, размером 2,4 ´ 2,3 ´ 1,5 см с незначительными явлениями перифокального отека. После в/в введения контраста в области вышеописанных изменений усиления интенсивности сигнала не отмечено. В целом отмечена отрицательная динамика по сравнению с предыдущими исследованиями. В связи с нарастающей общемозговой неврологической симптоматикой проведена КТ головного мозга от 26.09.2007 г. (Красноярск): на серии томограмм обнаружена «свежая кровь» в области намета мозжечка слева, в области цистерн мозжечка, в третьем желудочке и в передних рогах боковых желудочков; в области червя мозжечка слева обнаружена зона неоднородно повышенной плотности размером 1,2 ´ 1,5 см. Сделано заключение о продолженном росте опухоли мозжечка, осложненном субарахноидально-вентрикулярным кровоизлиянием. Порэнцефалия. Рубцово-атрофический процесс в области червя мозжечка. Дополнительно, по данным МРТ головного мозга (1,5Тс) с контрастированием гадолинием (омнискан в/в 10 мл) в сочетании с МРА церебральных сосудов от 25.10.2007 г. (Красноярск): отмечена отрицательная динамика в сравнении с предыдущим исследованием в виде рецидива объемного образования мозжечка с кровоизлиянием в опухоль, контуры опухоли четкие неровные, размер 2,9 ´ 3,0 ´ 2,3 см, с компрессией задних отделов пластинки четверохолмия и с умеренно выраженной перифокальной реакцией, с явлениями накопления контраста; в левой височной доле в субконвекситальных отделах выявлено объемное образование размером 2,4 ´ 2,3´1,5 см с четкими неровными контурами с явлениями незначительного перифокального отека, без явлений накопления контрастного вещества; выявлена умеренно выраженная внутренняя гидроцефалия. Вариант развития виллизиева круга в виде сужения просвета и снижения кровотока по левой задней соединительной артерии.
Ребенок проконсультирован детским хирургом ГБ № 20 им. Берзона (Красноярск). С учетом сложности локализации опухоли принято решение о направлении больного в НИИ нейрохирургии им. акад. Н.Н. Бурденко (Москва) для решения вопроса о хирургическом лечении или использовании методики «гамма-нож». В ноябре 2007 г. больному проведена ПЭТ головного мозга в Институте мозга РАН с введением радиофармпрепарата (РФП) 11С-метионина внутривенно, введенная доза составила 20,38 mCi 11С-метионина. Лучевая нагрузка на все тело при ПЭТ-исследовании составила 4,08 мЗв. На серии позитронно-эмиссионных томограмм 915 срезов в черве и оральном участке левого полушария мозжечка выявлен очаг слегка повышенного неоднородного накопления РФП (ИН = 1,24) размером 25 ´24´ ´34 мм. В центре описанного образования намечался гипометаболический участок неправильной формы. Локализация образования в целом совпадала с зоной контрастного усиления и кровоизлияния, обнаруженного на МРТ и КТ головного мозга. В левой височной доле очагов патологического накопления РФП не выявлено, поэтому сделано заключение о постлучевом кровоизлиянии в зону операции после комбинированного лечения пилоцитарной астроцитомы червя и полушария мозжечка. ПЭТ-признаков отсевов опухоли в височную долю не получено. Рекомендована ПЭТ через 6 месяцев.
КТ головного мозга проведена 12 ноября 2007 г. на базе нейрорентгенологического отделения НИИ им.акад. Н.Н. Бурденко (Москва): на серии контрольных компьютерных томограмм головного мозга в стандартном перфузионном режиме сканирования на фоне локального дефекта мозгового вещества в проекции верхнего червя мозжечка определялись небольшие остатки опухоли, расположенные по периферии указанного дефекта (лучше определяемые в передневерхних аспектах). Кроме того, имелся микропетрификат в остатках опухоли справа. Данных за выраженные изменения перфузии в остатках новообразования не выявлено. Обращало на себя внимание наличие дополнительных изменений в веществе мозга: в проекции моста, в подкорковых узлах с двух сторон, а также в проекции корковых отделов височной доли. Сделано заключение о состоянии после комбинированного лечения опухоли верхнего червя мозжечка с остатками опухоли без анапластической прогрессии, подозрение на нейрофиброматоз 1-го типа (болезнь Реклингхаузена) с наличием дизэмбриопластической нейроэпителиальной опухоли (или кортикальной дисплазии) в левой височной области. Рекомендована МРТ головного мозга с контрастированием гадолинием через 6 месяцев. 14 ноября 2007 г. ребенок вновь проконсультирован нейрохирургом Москвы. Клинический диагноз: продолженный рост пилоцитарной астроцитомы мозжечка, состояние после удаления опухоли (в июле 2004 г.) с применением компьютерной планирующей системы Leksell Gamma Plan Wizard 5.34, выполнено планирование сеанса радиохирургии. Проведен 1 сеанс. Общее количество точек цели составило 29. Объем мишени — 7,541 см3, размер мишени — 42´23 ´´ 26 мм. Предписанная доза— 18 Гр. Предписанная изодоза— 50%. Объем мишени, полученный предписанной дозой, — 6,819 см3. Минимальная доза — 11,52Гр. Максимальная доза — 36,00Гр. Общее время облучения составило 217 минут. Доза облучения в изоцентр составила 36 Гр. Объем черепа, облученный дозой более 10 Гр, — 39 см3. Интегральная доза на череп — 6 Дж. После окончания сеанса облучения рама снята, наложены асептические пластыри. Процедуру пациент перенес удовлетворительно. Введен внутривенно дексаметазон 16 мг. Рекомендовано: стероидная терапия по схеме, контрольная МРТ головного мозга с контрастированием с оптимальной толщиной МРТ-срезов 1 мм, максимальной — 5 мм через 6 мес., а также повторная консультация нейрохирурга через полгода.
Таким образом, пробанд неоднократно обследован и консультирован в нейрохирургических клиниках Москвы, однако клинический диагноз нейрофиброматоза 1-го типа (болезни Реклингхаузена), несмотря на наличие клинических и нейрорадиологических признаков, не был установлен. В дальнейшем (2007–2010гг.) ребенок состоял на диспансерном учете у нейрохирурга по месту жительства, однако на консультацию к нейрогенетику и неврологу не направлялся. Вновь проведена МРТ головного мозга (1,5 Тс) с контрастированием гадолинием (омнискан в/в 10 мл) 03.08.2008г.: выявлена незначительная положительная динамика (по сравнению с МРТ от 19.03.2008 г.) в виде уменьшения размеров объемного образования в области червя мозжечка по задней поверхности ствола мозга, в проекции пластинки четверохолмия. Структурная картина, с учетом изменений в базальных ядрах и стволе мозга (в проекции лентикулярных ядер и медиальных отделов зрительных бугров, в ножках мозжечка, в проекции зубчатых ядер мозжечка — в виде участков гиперинтенсивного сигнала по Т2 и в FLAIR-режиме и изогипоинтенсивного по Т1, размером 1,7 ´ 0,8 см без четких контуров (гамартомы?) могла соответствовать нейрофиброматозу 1-го типа. Размеры объемного образования в височной доле слева (нейроэпителиальные ядра?) — без отрицательной динамики по сравнению с предыдущими МРТ-исследованиями (рис. 1–3).

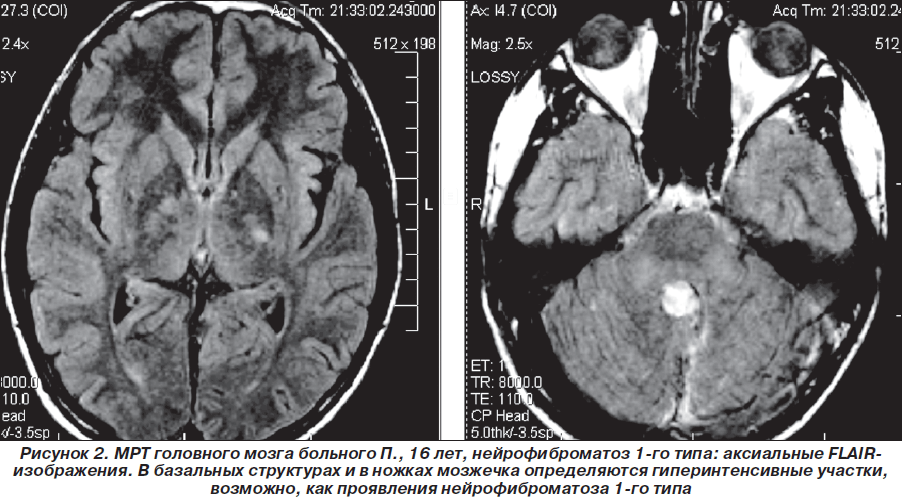
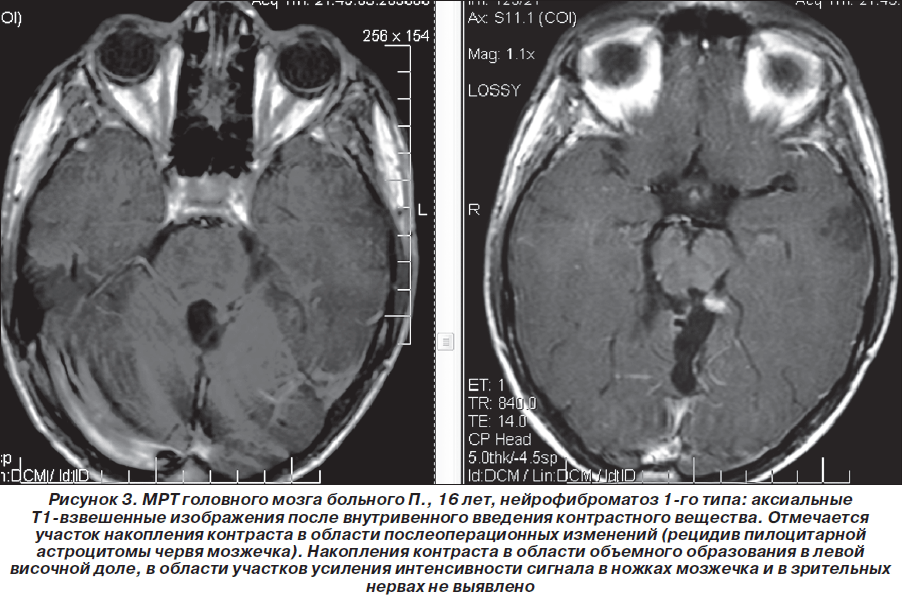
В августе 2008 г. больной повторно проконсультирован в нейрохирургическом центре Москвы. Несмотря на вышеизложенное, клинический диагноз остался прежним: пилоидная астроцитома мозжечка, состояние после удаления опухоли (2004 г.), состояние после стереотаксической радиохирургии на аппарате гамма-нож (14.11.2007 г.). Консультантом состояние больного расценено как удовлетворительное, признаки продолженного роста опухоли не вынесены в клинический диагноз. Рекомендована контрольная МРТ головного мозга с контрастированием через 1 год или раньше (при условии ухудшения состояния).
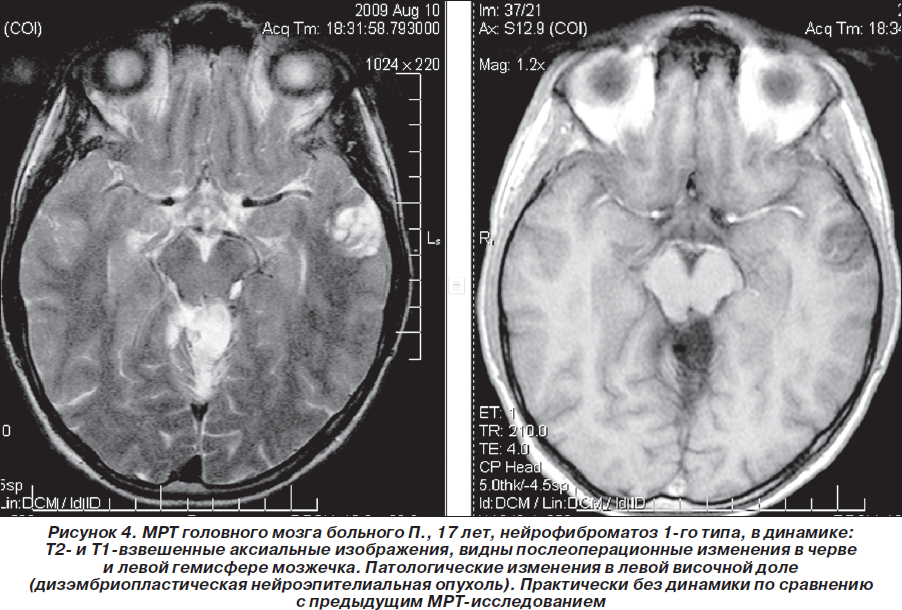
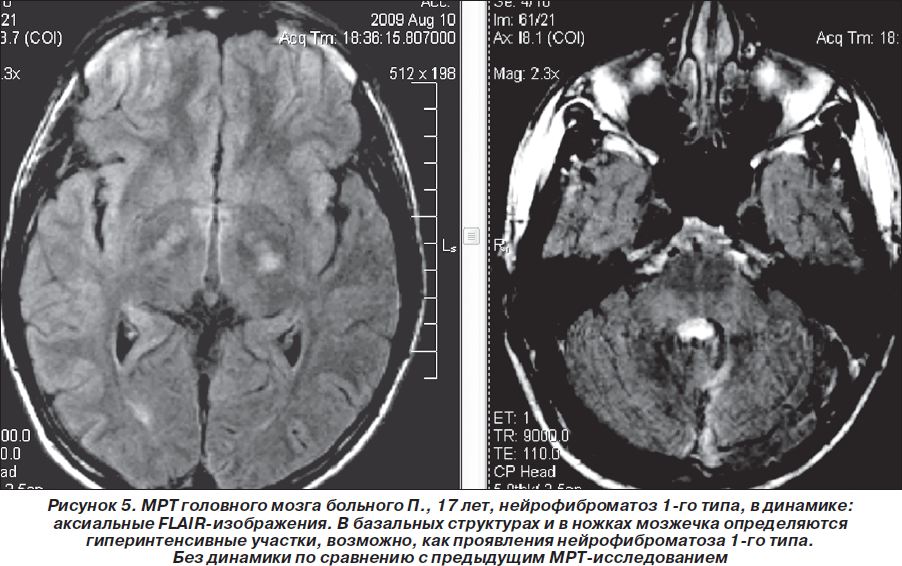
В мае 2009 г. повторно проведена МРТ головного мозга 1,5 Тс с контрастированием гадолинием (Красноярск): отчетливой отрицательной динамики в сравнении с предыдущей МРТ от 03.08.2008 г. не выявлено, объемное образование в области четверохолмной цистерны и левой гемисферы мозжечка не увеличилось в объеме, интенсивность его контрастирования несколько снизилась. Однако обращало на себя внимание объемное образование в височной доле левого полушария, которое структурно соответствовало дизэмбриопластической нейроэпителиальной опухоли, без тенденции к росту по сравнению с предыдущим исследованиями (размер 2,2 ´ 2,3 ´ 1,9 см), не исключались гамартомы в базальных структурах, ножках мозжечка и в проекции зубчатых ядер мозжечка. Сделано предположение о наличии у больного нейрофиброматоза 1-го типа. Нейрорадиологом МРТ-центра (Красноярск) впервые больному рекомендована консультация нейрогенетика (рис. 4–6). Но эта рекомендация была проигнорирована родителями ребенка — пробанд продолжал наблюдаться только у нейрохирурга по месту жительства. Рекомендовано радиолечение (гамма-нож).

В мае 2009 г. дано направление для освидетельствования на МСЭК по месту жительства в связи «с сохранением когнитивных и довольно выраженных координаторных нарушений». В мае того же года впервые в жизни во время ночного сна у ребенка развился вторично-генерализованный тонико-клонический припадок. Проведена рутинная ЭЭГ (15.11.2007 г.): выявлена полифокальная эпилептиформная активность с доминирующим фокусом в левой височной доле головного мозга с нарастанием ее мощности при двухминутной гипервентиляции с феноменом вторичной билатеральной синхронизации — трансформация фокальной эпилептиформной активности во вторично-генерализованную. В связи этим ребенок повторно проконсультирован нейрохирургом по месту жительства, но, несмотря на данные параклинических обследований и ярко выраженную клиническую неврологическую симптоматику, диагноз остался прежним, рекомендовано «диспансерное наблюдение у нейрохирурга, продление инвалидности и МРТ головного мозга через год». Консультация невролога-эпилептолога не рекомендована, противоэпилептические препараты не подобраны. В связи с рецидивом объемного образования и появлением новых очагов в головном мозге данная тактика ведения больного привела к медленному, но неуклонному прогрессированию симптоматической (онкогенной) фокальной эпилепсии с развитием в конце августа 2009 г. во время ночного сна повторного вторично-генерализованного гипермоторного припадка с фокальным компонентом в начале и в конце припадка в виде насильственного поворота головы и глаз влево, тонической установки верхних конечностей по типу позы «фехтовальщика». Однако ребенок и на этот раз не проконсультирован неврологом-эпилептологом, противоэпилептические препараты не назначены.
На фоне отсутствия противоэпилептической терапии симптоматической (онкогенной) фокальной эпилепсии в период с августа 2009 г. по январь 2010 г. участились простые фокальные адверсивные припадки в дневное время суток, в последующем участились вторично-генерализованные гипермоторные припадки (7 октября 2009 г., 8 января 2010 г., 19 января 2010 г., 20 января 2010 г.). Кроме того, после повторных курсов радиотерапии нарастали явления вторичного иммунодефицита с появлением гнойничковых высыпаний на коже и частыми «простудными» заболеваниями, однако на консультацию к иммунологу лечащим врачом нейрохирургом больной не направлялся.
Объективно (на момент настоящей консультации): при тщательном осмотре кожи ребенка на переднебоковой поверхности живота слева выявлено одиночное пятно цвета кофе с молоком с четкими неровными границами, листовидной формы, размером 18,0´7,5см, 17 пятен на коже спины, шеи, верхних и нижних конечностей размером от 0,5 ´ 0,8 см до 1,5 ´ 2,2 см. На спинке носа и на щеках отмечены мелкие ангиофибромы размером с просяное зерно (по типу «крыльев бабочки»). В неврологическом статусе: умеренные когнитивные нарушения динамического характера, умеренная статико-динамическая мозжечковая атаксия. Наследственный анамнез отягощен по материнской линии (рис. 7): у матери также имелись пятна цвета кофе с молоком на коже, однако уточнить их размеры и количество на момент настоящей консультации не представлялось возможным ввиду отсутствия матери пациента на приеме. Провести клинико-генеалогический анализ родословной по материнской линии на момент консультации также не представлялось возможным по тем же причинам. Пробанд — единственный ребенок в семье. Со слов отца больного, у матери пробанда привычное невынашивание беременности, женщина находилась на диспансерном наблюдении у акушера-гинеколога, проходила курсовое лечение, планировалось экстракорпоральное оплодотворение. Однако, несмотря на наличие кожных симптомов нейрофиброматоза 1-го типа (болезни Реклингхаузена), мать пробанда на предмет сочетанного поражения ЦНС ранее не обследовалась. На диспансерном учете у медицинского генетика в Красноярском консультативно-диагностическом центре медицинской генетики пробанд и его мать не состояли. Медико-генетическое консультирование семьи ранее не проводилось, несмотря на длительный анамнез заболевания у пробанда и его матери.
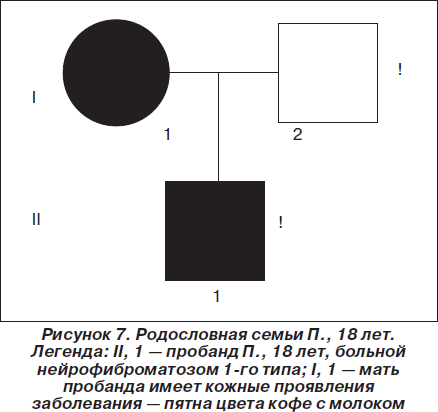
При отсутствии междисциплинарного подхода к ведению пробанда врачами различных специальностей (педиатрами, дерматологами, нейрохирургами, радиохирургами, нейрорадиологами, офтальмологами) клинический диагноз нейрофиброматоза 1-го типа (болезни Реклингхаузена) не был установлен в течение 18 лет. В 2009 г. нами впервые на основании жалоб, характерных клинических проявлений, данных параклинических исследований, отягощенного наследственного анамнеза впервые был сформулирован клинический диагноз: нейрокожный синдром (факоматоз), семейная форма, аутосомно-доминантный тип наследования: нейрофиброматоз 1-го типа (болезнь Реклингхаузена), впервые выявленный, с поражением центральной нервной системы (состояние после субтотальной резекции и радиотерапии пилоцитарной астроцитомы червя мозжечка в июле 2004г., послеоперационные кистозно-глиозно-атрофические изменения в области червя мозжечка, рецидив пилоцитарной астроцитомы с кровоизлиянием в ноябре 2007 г., с распространением в область четверохолмной цистерны и на пластинку четверохолмия, состояние после стереотаксической радиохирургии на аппарате гамма-нож в ноябре 2007г.; объемное образование левой височной доли; мелкие множественные гамартомы на уровне лентикулярных ядер и медиальных отделов зрительных бугров, в ножках мозжечка и в области зубчатых ядер мозжечка; умеренная мозжечковая статико-динамическая атаксия; умеренно выраженная преимущественно внутренняя асимметричная гидроцефалия; ликвородинамические кризы с умеренным краниалгическим синдромом; симптоматическая лобно-долевая эпилепсия с простыми и комплексными адверсивными и версивными фокальными припадками, одиночными вторично-генерализованными тонико-клоническими и гипермоторными припадками, дебют, некомпенсированная; умеренные когнитивные нарушения динамического характера); с поражением кожи (множественные пятна гиперпигментации на конечностях и туловище, мелкие ангиофибромы лица).
Сопутствующий диагноз: вторичный иммунодефицит после повторных сеансов радиотерапии. Рецидивирующий фурункулез, вне обострения. Хроническая дистальная вегетосенсорная полинейропатия нижних конечностей 1-й ст. тяжести, впервые выявленная.
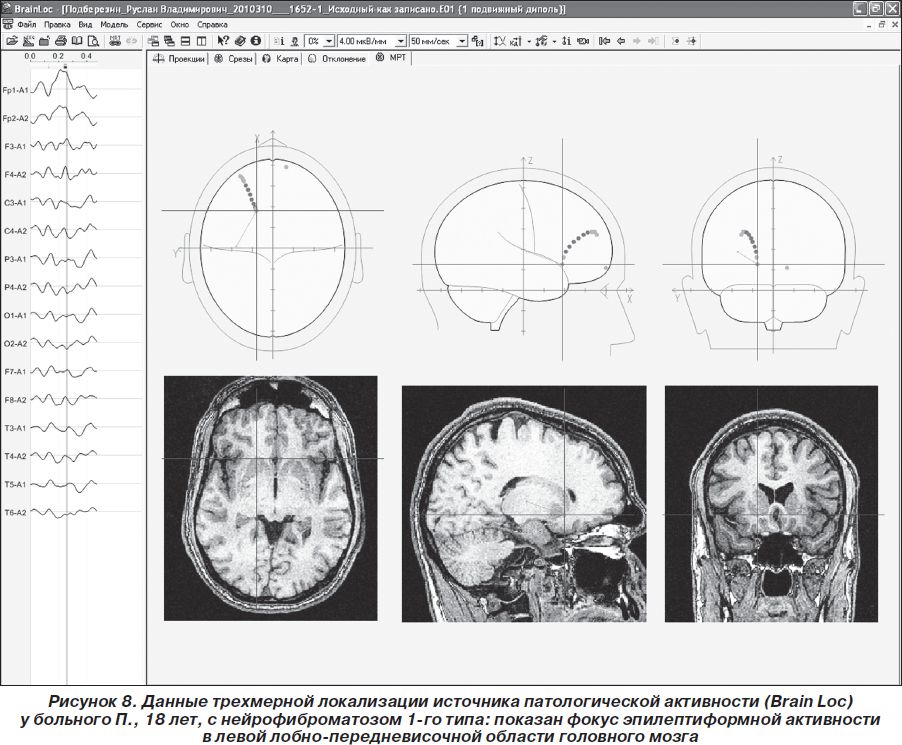
Пробанду рекомендован видео-ЭЭГ-мониторинг, подбор антиэпилептических препаратов (АЭП) и диспансерное наблюдение у невролога-эпилептолога, нейрогенетика и иммунолога. Комплайентность пробанда и членов его семьи к сотрудничеству с врачом-эпилептологом была низкой — пациент в назначенное время на повторный прием не явился, АЭП не получал. Спустя месяц после даты назначенного диспансерного приема пациент вновь обратился на консультацию в Неврологический центр эпилептологии, нейрогенетики и исследования мозга в связи с прогрессированием основного заболевания. Участились и стали протекать более длительно фокальные и вторично-генерализованные припадки, имелась тенденция к их серийному течению. По неотложным показаниям проведен видео-ЭЭГ-мониторинг (1 час): зарегистрирован доминирующий интериктальный фокус эпилептиформной активности в лобно-центральной области головного мозга слева, чувствительный к афферентным раздражителям (ритмической и триггерной фотостимуляции) и к гипоксии (рис. 8). Вновь настоятельно рекомендован прием АЭП, а также дообследование пробанда и его родственников. Однако в последующем (около 5 мес.) пациент на повторные осмотры к эпилептологу и нейрогенетику не обращался, до настоящего времени родственники пробанда 1-й и 2-й линии родства не дообследованы, что свидетельствует о крайне низкой комплайентности семьи пробанда к сотрудничеству с врачом, несмотря на ярко выраженную и непрерывно прогрессирующую симптоматику болезни Реклингхаузена.
Заключение
Раннее выявление NF1 на уровне педиатрической службы позволяет определить междисциплинарную тактику диспансерного наблюдения больного (пробанда), асимптомных (малосимптомных) и симптомных членов родословной — носителей мутантного гена. Важным аспектом медицинской помощи является медико-генетическое консультирование членов семьи, что способствует снижению риска рождения больных с тяжелыми формами NF1. Авторы подчеркивают важность повышения уровня профессиональной подготовки и настороженности врачей первичного звена здравоохранения в отношении выявления наиболее распространенных форм факоматозов, а также преемственности между специалистами различного профиля с целью своевременного проведения комплекса дополнительных методов диагностики и уменьшения степени инвалидизации пациентов трудоспособного возраста. Важно помнить, что процесс развития клинической симптоматики NF1 является динамическим— медленно, но непрерывно прогредиентным [35–40].
1. Аверьянов Ю.Н. Нейрокожные синдромы. — М.: Медицина, 2003. — С. 27-35.
2. Балязин В.А., Кравченко М.И., Фомина-Чертоусова Н.А. Нейрокожные синдромы: клиника, диагностика. — М.; Элиста: АПП «Джангар», 2001. — 96 с.
3. Шнайдер Н.А. Нейрофиброматоз 1-го типа: этиопатогенез, клиника, диагностика, прогноз // Международный неврологический журнал. — 2007. — № 5. — С. 162-168.
4. Friedman J.M., Birch P.H. Type 1 neurofibromatosis: a descriptive analysis of the disorder in 1,728 patients // Am. J. Med. Genet.— 1997. — Vol. 70. — P. 138-1434.
5. Griffiths P.D., Blaser S., Mukonoweshuro W. et al. Neurofibromatosis bright objects in children with neurofibromatosis type 1: a proliferative potential? // Pediatrics. — 1999. — Vol. 104. — P. 49.
6. Gutmann D.H. Recent insights into neurofibromatosis type1: clear genetic progress // Arch. Neurol. — 1998. — Vol. 55. — P.778-780.
7. Gutmann D.H., Aylsworth A., Carey J.C. et al. The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2 // JAMA. — 1997. — Vol. 278. — P. 51-57.
8. Allanson J.E., Upadhyaya M., Watson G.H. et al. Watson syndrome: is it a subtype of type 1 neurofibromatosis? // J. Med. Genet.— 1991. — Vol. 28. — P. 752-756.
9. Barton B., North K. Social skills of children with neurofibromatosis type 1 // Dev. Med. Child. Neurol. — 2004. — Vol. 46. — P.553-563.
10. Zhu Y., Parada L.F. Neurofibromin, a tumor suppressor in the nervous system // Exp. Cell Res. — 2001. — Vol. 264. — P. 19-28.
11. Cichowski K., Jacks T. NF1 tumor suppressor gene function: narrowing the GAP // Cell. — 2001. — Vol. 104. — P. 593-604.
12. Clementi M., Milani S., Mammi I. et al. Neurofibromatosis type 1 growth charts // Am. J. Med. Genet. — 1999. — Vol. 87. — P.317-323.
13. Gutmann D.H. Recent insights into neurofibromatosis type1: clear genetic progress // Arch. Neurol. — 1998. — Vol. 55. — P.778-780.
14. Lothe R.A., Slettan A., Saeter G. et al. Alterations at chromosome 17 loci in peripheral nerve sheath tumors // J. Neuropathol. Exp. Neurol. — 1995. — Vol. 54. — P. 65-73.
15. Shen M.H., Harper P.S., Upadhyaya M. Molecular genetics of neurofibromatosis type 1 (NF1) // J. Med. Genet. — 1996. — Vol.33.— P. 2-17.
16. Rutkowski J.L., Wu K., Gutmann D.H. et al. Genetic and cellular defects contributing to benign tumor formation in neurofibromatosis type 1 // Hum. Mol. Genet. — 2000. — Vol. 9. — P. 1059-1066.
17. Шнайдер Н.А., Горелов А.И. Нейрофиброматоз I типа: болезнь Реклинхаузена // Сибирское медицинское обозрение. — 2007. — Т. 44, № 3. — С. 91-95.
18. Evans D.G., Baser M.E., McGaughran J. et al. Malignant peripheral nerve sheath tumours in neurofibromatosis 1 // J. Med. Genet. — 2002. — Vol. 39. — P. 311-314.
19. Friedman J.M., Birch P.H. Type 1 neurofibromatosis: a descriptive analysis of the disorder in 1,728 patients // Am. J. Med. Genet. — 1997. — Vol. 70. — P. 138-143.
20. Korf B.R. Malignancy in neurofibromatosis type 1 // Oncologist. — 2000. — Vol. 5. — P. 477-485.
21. Tada K., Kochi M., Saya H. et al. Preliminary observations on genetic alterations in pilocytic astrocytomas associated with neurofibromatosis 1 // Neuro-oncol. — 2003. — Vol. 5. — P. 228-234.
22. Trimbath J.D., Petersen G.M., Erdman S.H. et al. Cafe-au-lait spots and early onset colorectal neoplasia: a variant of HNPCC?// Fam. Cancer. — 2001. — Vol. 1. — P. 101-105.
23. Listernick R., Mancini A.J., Charrow J. Segmental neurofibromatosis in childhood // Am. J. Med. Genet. — 2003. — Vol.121A.— P.132-135.
24. Lothe R.A., Slettan A., Saeter G. et al. Alterations at chromosome 17 loci in peripheral nerve sheath tumors // J. Neuropathol. Exp. Neurol. — 1995. — Vol. 54. — P. 65-73.
25. Molloy P.T., Bilaniuk L.T., Vaughan S.N. et al. Brainstem tumors in patients with neurofibromatosis type 1: a distinct clinical entity // Neurology. — 1995. — Vol. 45. — P. 1897-1902.
26. Vinchon M., Soto-Ares G., Ruchoux M.M., Dhellemmes P. Cerebellar gliomas in children with NF1: pathology and surgery // Childs Nerv. Syst. — 2000. — Vol. 16. — P. 417-420.
27. Ricciardone M.D., Ozcelik T., Cevher B. et al. Human MLH1 deficiency predisposes to hematological malignancy and neurofibromatosis type 1 // Cancer Res. — 1999. — Vol. 59. — P. 290-293.
28. Rosser T.L., Packer R.J. Neurocognitive dysfunction in children with neurofibromatosis type 1 // Curr. Neurol. Neurosci. Rep.— 2003. — Vol. 3. — P. 129-136.
29. DeBella K., Poskitt K., Szudek J., Friedman J.M. Use of «unidentified bright objects» on MRI for diagnosis of neurofibromatosis 1 in children // Neurology. — 2000. — Vol. 54. — P. 1646-1651.
30. De Bella K., Szudek J., Friedman J.M. Use of the national institutes of health criteria for diagnosis of neurofibromatosis 1 in children // Pediatrics. — 2000. — Vol. 105. — P. 608-614.
31. Di Paolo D.P., Zimmerman R.A., Rorke L.B. et al. Neurofibromatosis type 1: pathologic substrate of high-signal-intensity foci in the brain // Radiology. — 1995. — Vol. 195. — P. 721-724.
32. Buehning L., Curry C.J. Neurofibromatosis-Noonan syndrome// Pediatr. Dermatol. — 1995. — Vol. 12. — P. 267-271.
33. Carey J.C. Neurofibromatosis-Noonan syndrome // Am. J. Med. Genet. — 1998. — Vol. 75. — P. 263-264.
34. De Bella K., Szudek J., Friedman J.M. Use of the national institutes of health criteria for diagnosis of neurofibromatosis 1 in children // Pediatrics. — 2000. — Vol. 105. — P. 608-614.
35. Шнайдер Н.А. Нейрофиброматоз 1 типа (болезнь Реклингхаузена): Учебное пособие для системы последипломного образования врачей. — КрасГМА, 2008. — 102 с.
36. Drouin V., Marret S., Petitcolas J. et al. Prenatal ultrasound abnormalities in a patient with generalized neurofibromatosis type 1// Neuropediatrics. — 1997. — Vol. 28. — P. 120-121.
37. Dugoff L., Sujansky E. Neurofibromatosis type 1 and pregnancy// Am. J. Med. Genet. — 1996. — Vol. 66. — P. 7-10.
38. Szudek J., Birch P., Riccardi V.M. et al. Associations of clinical features in neurofibromatosis 1 (NF1) // Genet. Epidemiol. — 2000.— Vol. 19. — P. 429-439.
39. Tonsgard J.H., Kwak S.M., Short M.P., Dachman A.H. CT imaging in adults with neurofibromatosis-1: frequent asymptomatic plexiform lesions // Neurology. — 1998. — Vol. 50. — P. 1755-1760.
40. Virdis R., Street M.E., Bandello M.A. et al. Growth and pubertal disorders in neurofibromatosis type 1 // J. Pediatr. Endocrinol. Meta