
Международный эндокринологический журнал 3 (35) 2011
Вернуться к номеру
NF-kB-сигнализация как основа развития системного воспаления, инсулинорезистентности, липотоксичности, сахарного диабета 2-го типа и атеросклероза
Авторы: Кайдашев И.П., НИИ генетических и иммунологических основ развития патологии и фармакогенетики Высшего государственного учебного заведения «Украинская медицинская стоматологическая академия», г. Полтава
Рубрики: Эндокринология
Версия для печати
В данной работе анализируются современные представления о нарушении регуляции активности NF-kB. Ядерный фактор NF-kB является мощным транскрипционным фактором, активирующим большинство провоспалительных механизмов в патогенезе атеросклероза. Свойство NF-kB ингибировать передачу инсулинового сигнала обусловлено его способностью вызывать фосфорилирование IRS-1 по остаткам серина.
Впервые выдвинуто предположение, что нарушение NF-kB-сигнализации может быть тем общим звеном, которое объединяет все компоненты метаболического синдрома и приводит к инсулинорезистентности, липотоксичности, системному воспалению, артериальной гипертензии. Влияния, которые приводят к снижению транскрипционной активности NF-kB (диета, физические нагрузки, медикаментозные препараты), будут модулировать состояние прекондиционирования, которое устраняет молекулярную основу развития метаболического/инсулинорезистентного синдрома и снижает риск сердечно-сосудистых заболеваний.
Сахарный диабет, инсулинорезистентность, NF-kB-сигнализация.
Сахарный диабет (СД) 2-го типа является комплексным метаболическим нарушением, сопровождающимся микро- и макрососудистыми нарушениями [19]. Гипергликемия является основным фактором риска развития микрососудистых нарушений, снижение уровня гликозилированного гемоглобина (HbA1c) уменьшает частоту развития ретинопатии, нефро- и нейропатии [11]. Макрососудистые осложнения являются первичной причиной смерти от инфаркта миокарда и инсульта у 80 % таких пациентов [50]. Частота основных атеросклеротических осложнений у пациентов с CД 2-го типа с предшествующим сердечно-сосудистым событием очень высока — около 6 % в год [59]. Проведенные современные исследования, суммированные и проанализированные в работе R.A. DeFronzo (2010), позволяют объяснить связь между высоким уровнем сердечно-сосудистых заболеваний (ССЗ) у пациентов с CД 2-го типа и явлениями инсулинорезистентности и липотоксичности [20]. Тем не менее эти синдромы, по-нашему мнению, являются лишь клиническими проявлениями важных нарушений, происходящих в системе NF-kB-сигнализации. Ниже мы приведем основные литературные данные и результаты проведенных нами исследований, обосновывающих концепцию прекондиционирования системы NF-kB-сигнализации с развитием системного воспаления у больных CД 2-го типа, ишемической болезнью сердца (ИБС), метаболическим синдромом (МС).
Гипергликемия, инсулин, атеросклероз и сердечно-сосудистые заболевания
Проведенные клинические исследования АССORD, ADVANCE и VADТ подняли важный вопрос о роли гипергликемии в развитии ССЗ [26, 34, 43]. Отсроченные наблюдения тем не менее показали значительное снижение частоты атеросклеротических сердечно-сосудистых событий [7]. Эти данные предполагают существование «эффекта памяти», улучшенного контроля гликемии, напоминающего исследование контроля диабета и осложнений у больных CД 1-го типа [45].
Анализ этих исследований показывает, что они в определенной степени не учитывали фундаментальные патофизиологические доказательства. Гипергликемия является достаточно слабым фактором риска СС3 в сравнении с такими хорошо доказанными факторами, как дислипидемия и гипертензия, более того, большинство больных в этих исследованиях получали комплексную терапию с гиполипидемическими, антигипертензивными и противовоспалительными препаратами. Пациенты достаточно долгое время страдали СД 2-го типа с предшествующими сердечно-сосудистыми событиями или многочисленными факторами риска СС3. Применение инсулина может быть не оптимальным методом для предотвращения ССЗ. Инсулинотерапия может вызывать прибавку веса, а прибавка веса (ожирение) является важным фактором риска СС3 [9, 10]. Более того, даже незначительное увеличение сывороточной концентрации инсулина может вызывать тяжелую инсулинорезистентность, являющуюся фактором риска СС3 [53].
Перечисленные выше доводы показывают, что гипергликемия, по сути, является отражением инсулинорезистентности и гиперинсулинемии и именно в этих явлениях может заключаться риск СС3.
Значительное число исследований in vivo и in vitro показало, что инсулин, особенно применяемый в высоких дозах, может интенсифицировать процессы атерогенеза [21]. Основные свойства инсулина при этом заключаются:
— в усилении синтеза липопротеидов очень низкой плотности (ЛПОНП) и возможном снижении уровня холестерина липопротеидов высокой плотности (ЛПВП);
— повышении транспорта холестерина в клетки гладкой мускулатуры артериол;
— активности, присущей фактору роста, — увеличении синтеза коллагена, стимуляции деления гладкомышечных клеток артериол;
— усилении атерогенеза в моделях на животных разных видов;
— индукции инсулинорезистентности.
Инсулин способен стимулировать липогенез и усиливать синтез ЛПОНП в печени путем активации белка 1с, связывающего стеролрегулирующий элемент, и ингибирование ацетил-СоА-карбоксилазы [39]. Инсулин является не только мощным ростовым фактором, стимулирующим образование коллагена, пролиферацию гладкомышечных клеток, но и, что особенно важно в рамках предлагаемой концепции, включает множество генов, определяющих активацию воспаления [25]. Важным также является исследование, показавшее, что у крыс, которым на протяжении 7–10 дней вводили инсулин, на фоне эугликемии развивалась резистентность к стимуляции захвата глюкозы, снижение уровня неэстерифицированных жирных кислот (НЭЖК) в плазме крови и возникала гипертензия [49]. У людей с нормальной чувствительностью к глюкозе введение инсулина в течение 3 дней приводит к увеличению уровня инсулина в плазме натощак с 57 до 104 пкмоль/л, вызывая значительную инсулинорезистентность (фактор риска СС3) [38]. Кроме того, активный контроль концентрации HbAc1 с помощью инсулина приводит к повышению массы тела, например снижение HbAc1 с 7,7 до 5,1 % привело к увеличению веса на 8,6 кг [44]. Набор веса, в свою очередь, стимулирует атерогенез с участием дислипидемии и гипертензии, накопление липидов в сосудистой стенке усиливает процессы воспаления, что напрямую активирует атерогенез [70].
Синдром инсулинорезистентности (метаболический синдром)
В современной литературе приведено множество доказательств того, что МС приводит к развитию ССЗ. Показано, что пациенты с СД 2-го типа и сниженной массой тела, а также пациенты с ожирением и нормальной толерантностью к глюкозе являются резистентными к инсулину и такая инсулинорезистентность прямо воздействует на синтез гликогена [12, 57]. Основными компонентами, которые формируют синдром инсулинорезистентности, являются [3]:
— ожирение (особенно висцеральное);
— сниженная толерантность к глюкозе (нарушенная толерантность к глюкозе, изменение уровня глюкозы натощак, СД 2-го типа);
— гипертензия;
— дислипидемия (высокий уровень триглицеридов, низкие ЛПВП, малые ЛПНП-частицы);
— эндотелиальная дисфункция;
— атеросклеротические ССЗ;
— гиперинсулинемия;
— инсулинорезистентность.
Общим звеном, объединяющим все компоненты МС, является основная клеточно-молекулярная причина, приводящая к инсулинорезистентности, которая не только индуцирует системное воспаление и атерогенез, но и вызывает (или усиливает) другие компоненты МС. Такой причиной, по-нашему мнению, является нарушение NF-kB-сигнализации.
Гипертензия является одним из основных компонентов МС. Во многих исследованиях показано, что пациенты с гипертонией имеют инсулинорезистентность с нарушением синтеза гликогена. Дислипидемия, возникающая у пациентов с СД 2-го типа и пациентов с ожирением, является также одним из основных факторов риска ССЗ, при этом гипертриацилглицеринемия, но не гиперхолестеринемия связана с инсулинорезистентностью [47]. Пациенты, страдающие ИБС, имеют отчетливую инсулинорезистентность с нарушением синтеза гликогена в скелетной мускулатуре и резистентны к действию инсулина. С помощью позитронной эмиссионной томографии показано, что миокард пациентов с ИБС без СД 2-го типа и пациентов с ИБС и СД 2-го типа резистентен к действию инсулина [38, 63]. Инсулинорезистентность присутствует уже на стадии нарушенной толерантности к глюкозе и даже раннего обнаружения нарушения толерантности к глюкозе [68]. Такие соображения позволили R.A. De Fronzo (2007) назвать метаболический синдром синдромом инсулинорезистентности, что, по его мнению, отражает этиологию лучше, чем просто неопределенный кластер факторов, которые могут иметь отношение к патогенезу, а могут не иметь [46]. Однако, разделяя общие идеи с предложенной трактовкой, ниже мы приведем ряд доводов в пользу дальнейших поисков и определения ключевых молекулярных каскадов, лежащих в основе описанных нарушений, в частности нарушений NF-kB-сигнализации.
Молекулярные причины инсулинорезистентности и МС как предикторы ССЗ
Многие проведенные перспективные исследования показали, что инсулинорезистентность является предиктором развития ССЗ. Наличие МС увеличивает втрое риск развития ССЗ [16]. Четкая зависимость между HOMAIR и толщиной интимы и медии каротид была показана в работе B. Bedblad [60].
Для осуществления своих физиологических эффектов инсулин должен связаться со своими специфическими рецепторами на клеточной поверхности. Такое связывание активирует систему вторичных посредников, которые инициируют каскад фосфорилирования/дефосфорилирования, что стимулирует транспорт глюкозы (через GLUT4), фосфорилирование глюкозы (через гексокиназу ІІ), гликогенсинтетазу (которая контролирует синтез гликогена), фосфофруктокиназу и пируватдегидрогеназу (которые регулируют гликолиз и окисление глюкозы) [51] (рис. 1).
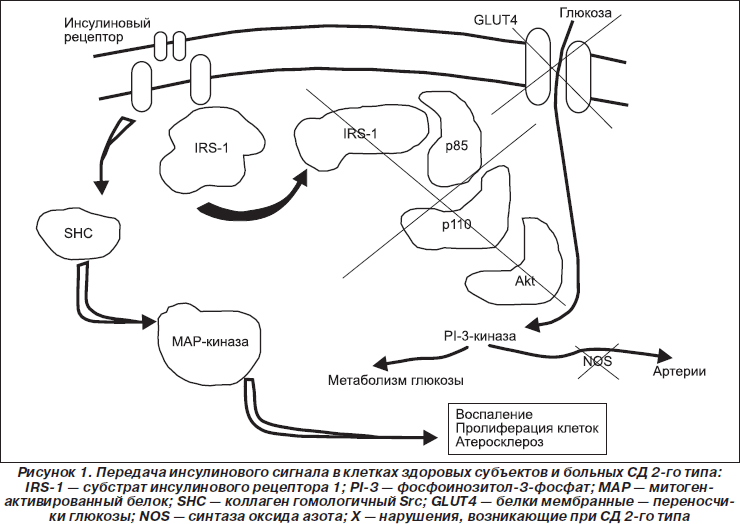
В мышцах инсулин связывается со своим рецептором, что приводит к фосфорилированию по тирозину IRS-1, который опосредует влияние инсулина на обмен глюкозы. В тканях печени эффекты инсулина опосредуются IRS-2. IRS-1 активирует фосфатидилинозитол-(РI)-3-киназу, которая катализирует 3’-фосфорилирование РI, РI-4-фосфата и РI-4,5-дифосфата и усиливает транспорт глюкозы и активность гликогенсинтетазы [15]. Ингибиторы РI-3-киназы ингибируют транспорт глюкозы, гексокиназу ІІ и гликогенсинтетазу.
Сигнальный путь инсулина играет важную роль в активации синтазы оксида азота, которая регулирует продукцию оксида азота [42]. Оксид азота является мощным вазодилататором и антиатеросклеротическим фактором, дефицит оксида азота активирует многие механизмы атерогенеза [48]. Таким образом, нарушения сигнального пути инсулина могут не только приводить к поломкам утилизации глюкозы, но и быть причиной гипертензии и ускорять прогрессирование атеросклероза.
Инсулин представляет собой достаточно сильный ростовой фактор, чьи эффекты опосредованы через МАР-киназный путь (mitogen-activated protein — МАР). После взаимодействия между IRS-1 и SHC активируется внеклеточно регулируемая киназа (ERK), переносится в ядро и катализирует фосфорилирование транскрипционных факторов, которые усиливают рост клеток, их пролиферацию и дифференцировку [66]. Приведенные сведения показывают, что этот путь играет важную роль в патогенезе атеросклероза. Блокада МАР-киназы предотвращает стимуляцию рост-усиливающих эффектов инсулина, но не оказывает действия на метаболическую активность инсулина [17]. Особо важными с позиций предлагаемой нами концепции являются данные о том, что инсулинорезистентность в РI-3-киназном (метаболическом) пути при неповрежденном МАР-киназном пути активирует множественные механизмы воспаления, включая ингибитор kВ (ІkВ) и ядерный фактор kВ (NF-kB) [61], и с-Jun N-концевую киназу, которые также могут вызывать инсулинорезистентность. Неудивительно, что значительная физиологическая гиперинсулинемия активирует многие гены, продукты которых участвуют в развитии воспаления [25] (рис. 2).
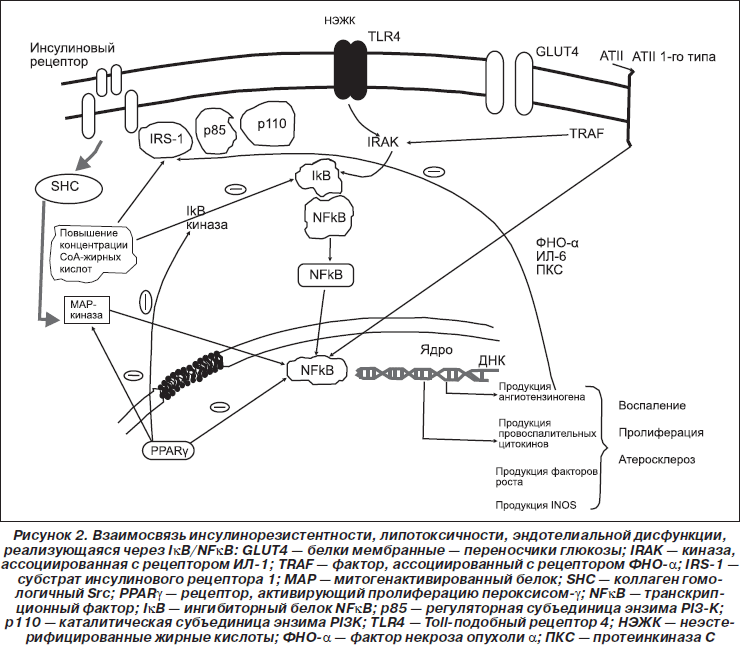
В литературе активно обсуждается вопрос о существовании дефектов в рецепторе инсулина при СД 2-го типа. Действительно показано снижение связывания инсулина с моноцитами и адипоцитами у больных СД 2-го типа. Тем не менее в мышечной ткани и печени связывание инсулина с солюбилизированными рецепторами вполне нормально как у полных людей с нормальной толерантностью к глюкозе, так и у больных СД 2-го типа с низким весом [40]. У больных СД 2-го типа с нормальным и избыточным весом тирозинкиназная активность инсулинового рецептора, стимулированная инсулином, снижена. Нормализация уровня глюкозы натощак и снижение массы тела приводили к нормализации тирозиновой киназы инсулинового рецептора, что свидетельствует о приобретенной природе этого нарушения [62].
Молекулярные основы связи инсулинорезистентности, липотоксичности и атеросклероза
Были получены данные, показывающие, что нарушенная активация фосфорилирования по тирозину IRS-1 и РI-3-киназы у больных СД 2-го типа с низким весом и лиц с нормальной толерантностью к глюкозе, но с избыточным весом, вызывает глубокие повреждения в транспорте и фосфорилировании глюкозы и синтезе гликогена [41]. Вследствие того что синтаза оксида азота активируется тем же РI-3-киназным путем, нарушается продукция оксида азота, что приводит к развитию эндотелиальной дисфункции и атеросклероза [14]. Это является упрощенной моделью молекулярной связи между инсулинорезистентностью, воспалением и развитием атеросклероза у больных СД 2-го типа.
Однажды возникший и закрепившийся дефект в сигнальном пути инсулина инициирует повторяющиеся циклы обратной связи. Нарушение утилизации глюкозы вызывает гипергликемию, которая стимулирует секрецию инсулина. Вследствие дефектности пути IRS-1/РI-3-киназы избыточно стимулируется МАР-киназный путь, так как он сохраняет нормальную чувствительность к инсулину. Повышенное фосфорилирование IRS-1 по серину, вызванное различными метаболическими и воспалительными нарушениями, обнаружено у больных СД 2-го типа [24], что еще больше повреждает сигнал от инсулина через РI-3-киназный путь. У больных СД 2-го типа и лиц с избыточным весом продолжающаяся стимуляция МАР-киназного пути увеличивает продукцию коллагена, вызывает пролиферацию гладкой мускулатуры сосудов, избыточное количество ростовых факторов и провоспалительных цитокинов, что усиливает развитие атеросклероза. Необходимо отметить, что большинство провоспалительных цитокинов, в свою очередь, усиливают экспрессию NF-kB и вызывают реципрокную активацию своего синтеза [31, 37].
Получены дополнительные доказательства такого двойственного эффекта инсулина. Культивируемые гладкомышечные клетки сосудов и эндотелиальные клетки, обработанные ингибиторами РI-3-киназы, не отвечали на воздействие инсулином путем активации синтазы оксида азота и не были защищены от повреждающих эффектов фактора роста сосудистого эпителия, ростового фактора тромбоцитов и других провоспалительных пептидов. Инсулин продолжал в этих условиях стимулировать пролиферацию и миграцию гладкомышечных клеток сосудов даже при ингибировании РI-3-киназы и усиливал экспрессию пренилированного Ras (онкогенный белок саркомы крыс) и Rho (гомолог Ras), приводя к усиленному ответу клеток на ростовой стимул от IGF-1, эпидермального фактора роста, ростового фактора тромбоцитов и ангиотензина ІІ [36].
В связи с основными положениями предлагаемой концепции сенситизация гладкомышечных клеток сосудов к ангиотензину ІІ является очень важной, так как гиперинсулинемия усиливает способность ангиотензина ІІ активировать NF-kB [35], при этом важный вклад может вносить полиморфизм гена рецептора ангиотензина ІІ 1-го типа [6]. Ядерный фактор NF-kB является мощным транскрипционным фактором, активирующим многие провоспалительные механизмы в патогенезе атеросклероза [52]. NF-kB также вызывает фосфорилирование IRS-1 по остаткам серина, что ингибирует передачу инсулинового сигнала [64].
Следует отметить, что ангиотензин ІІ также способен фосфорилировать IRS-1 в гладкой мускулатуре аорты и клетках скелетных мышц. Таким образом, каскад реакций, связанных с NF-kB, является связующим звеном между инсулинорезистентностью, атерогенезом и эссенциальной гипертензией.
Термин «липотоксичность» был введен Under для обозначения различных эффектов накопления жировой ткани на обмен глюкозы [73]. Экспериментальное увеличение концентрации неэстерифицированных жирных кислот до уровня характерного для больных СД 2-го типа вызывало тяжелую инсулинорезистентность в мышечной ткани и печени, ингибировало секрецию инсулина, по сути, вызывая основные признаки СД 2-го типа. Повышенные уровни НЭЖК нарушают окисление глюкозы, синтез гликогена, снижают транспорт и фосфорилирование глюкозы. Инфузия липидов, повышающая уровень НЭЖК, у пациентов с нормальной толерантностью к глюкозе вызывала дозозависимое торможение фосфорилирования инсулинового рецептора и IRS-1 по остаткам тирозина, снижение активности РI-киназы [23].
Внутриклеточные токсические метаболиты триацилглицерола и метаболиты НЭЖК (жирные кислоты, активированные СоА, диацилглицерол, церамиды) вызывают тяжелую инсулинорезистентность вследствие нарушения передачи инсулинового сигнала и множества стадий обмена глюкозы. Показано, что снижение уровня НЭЖК у больных СД 2-го типа с помощью ингибитора липолиза аципимокса с 563 до 230 ммоль/л приводило к снижению концентрации длинноцепочечных жирных кислот, активированных СоА, что сопровождалось усилением захвата глюкозы мышечной тканью под действием инсулина [28].
В исследованиях на животных и наблюдениях за больными было показано, что увеличение концентрации НЭЖК в плазме вследствие инфузии липидов приводит к увеличению в миоцитах уровня диацилглицерола, мощного активатора изоформ протеинкиназы С, которые ингибируют передачу инсулинового сигнала через фосфорилирование IRS-1 по остаткам серина. У лиц с ожирением и у больных СД 2-го типа также повышен уровень церамидов в мышечной ткани и в плазме, что коррелирует с тяжестью инсулинорезистент-ности.
НЭЖК, адипозопатия, воспаление и атеросклероз
Течение СД 2-го типа и ожирения можно охарактеризовать как низкоинтенсивное хроническое воспаление, которое определяет развитие атеросклероза [33]. Повышенная активность NF-kB представляет собой молекулярный механизм, ответственный за развитие воспаления и инсулинорезистентности при СД 2-го типа [65]. В базальных условиях NF-kB связан в цитоплазме со своим ингибитором IkB. При активации воспалительными факторами (активированные СоА жирные кислоты) IkB-киназа фосфорилирует IkB, вызывая полиубиквитинирование IkB и его деградацию. Свободный NF-kB транслоцируется в ядро, где присоединяется к гену-мишени, стимулируя продукцию медиаторов воспаления (ФНО-a, ИЛ-1b, ИЛ-6, ПКС), участвующих в атерогенезе [75]. ФНО-a, ИЛ-6 и ПКС, так же как ІК-киназа, вызывают фосфорилирование IRS-1 по остаткам серина, ингибируя инсулиновый сигнал и приводя к инсулинорезистентности [72].
При СД 2-го типа содержание IkB в скелетных мышцах больных снижено, что отражает повышение активности NF-kB. Множество стимулирующих факторов, в т.ч. липиды, активируют сигнальный путь NF-kB, вызывая состояние инсулинорезистентности в миоцитах организмов различных видов. В миоцитах человека пальмитат усиливает активность NF-kB и увеличивает образование мРНК ИЛ-6. В этих условиях трансфекция миоцитов суперрепрессорным мутантом аденовирус-IkB блокирует увеличение активности NF-kB и экспрессию мРНК ИЛ-6 под действием пальмитата [29].
Циркулирующие НЭЖК также способны вызывать развитие воспаления и инсулинорезистентности путем прямой активации TLR4. В мышцах лиц с ожирением и больных СД 2-го типа повышено содержание мРНК и белка TLR4, это тесно коррелирует с индексом инсулинорезистентности НОМА-IR [29] и активацией NF-kB, что обеспечивает еще один путь реализации провоспалительного эффекта НЭЖК и усиливает развитие атеросклероза.
НЭЖК также стимулируют образование коллагена, интегрального компонента атеросклеротической бляшки [18]. Физиологический подъем концентрации НЭЖК у лиц с нормальной толерантностью к глюкозе увеличивает экспрессию мРНК-коллагена, фибронектина и люмикана подобно фактору роста соединительной ткани, который обеспечивает фиброзный эффект. Экспериментальное увеличение плазменной концентрации НЭЖК у лиц с нормальной толерантностью к глюкозе снижает эндотелийзависимый кровоток без нарушения эндотелийнезависимого кровотока.
Адипозопатия представляет собой другую форму липотоксичности. Жировая ткань синтезирует адипокины, транспортирующиеся в отдаленные ткани (мышцы, печень, артерии), где они оказывают влияние на метаболизм и функциональное состояние сосудов. Жировая ткань пациентов с СД 2-го типа и лиц с ожирением инфильтрирована мононуклеарными клетками и находится в состоянии хронического воспаления [74]. Адипоциты и мононуклеарные клетки, инфильтрирующие жировую ткань, секретируют провоспалительные и тромботические цитокины (ФНО-a, резистин, ИЛ-6, ингибитор активатора плазминогена-1, ангиотензиноген и т.д.), которые усиливают процессы атерогенеза и вызывают инсулинорезистентность. Адипоциты таких больных продуцируют сниженное количество адипонектина — мощного инсулинсенситайзерного и антиатерогенного цитокина [13].
Во многих проведенных современных исследованиях показано, что накопление жировой ткани внутри брюшной полости (висцеральный жир) связано с развитием инсулинорезистентности и ССЗ [13]. Несмотря на то что механизмы накопления висцерального жира остаются до конца не изученными, такое висцеральное ожирение является составной частью метаболического синдрома.
Накопление жиров в тканях объясняется:
— поступлением в организм избыточной энергии;
— повышенным липолизом в инсулинорезистентных адипоцитах, который приводит к повышению уровня НЭЖК в плазме, и усиленным накоплением их в мышцах и печени;
— снижением окисления жиров.
В такой ситуации снижение веса (ограничение поступающей в организм энергии) оказывается эффективным средством уменьшения общего содержания жира в организме. Аналоги глюкагоноподобного пептида-1, угнетая аппетит, эффективно воздействуют на снижение массы тела [71].
Тиазолидиндионы оказывают уникальное действие по снижению липотоксичности:
— снижают уровень НЭЖК в плазме путем ингибирования липолиза;
— снижают концентрацию длинноцепочечных жирных кислот, активированных СоА, в мышцах;
— перераспределяют отложение жира в организме;
— снижают тяжесть адипозопатии.
Тиазолидиндионы являются мощными инсулиносенситайзерами в тканях мышц и печени, улучшают чувствительность к антилиполитическому действию инсулина, снижают базальный обмен НЭЖК и их концентрацию в плазме [27]. Пиоглитазон также способен снижать липотоксичность через адипонектин и его рецептор. Гипоадипонектинемия является характерным признаком СД 2-го типа. Тиазолидиндионы способны повышать уровень адипонектина до значений, свойственных здоровым людям. У больных СД 2-го типа и лиц с инсулинорезистентностью снижена экспрессия адипонектиновых рецепторов 1 и 2. Пиоглитазон усиливает экспрессию этих рецепторов в мышцах и АМР-киназы. Препарат также активирует ацетил-СоА-карбоксилазу и карнитинпальмитоилтрансферазу с окончательным ингибированием малонил-СоА. Это приводит к увеличению активности карнитинпальмитоилтрансферазы І и потока НЭЖК в митохондрии, где они подвергаются b-окислению. В результате этого снижается уровень жирных кислот, связанных с СоА в миоцитах, что улучшает передачу инсулинового сигнала и чувствительность к инсулину.
Активация PPARg пиоглитазоном приводит к снижению экспрессии NF-kB в CD40+-лимфоцитах [67].
Важными являются данные, что активация PPARg ингибирует TLR-опосредованную активацию NF-kB путем блокирования МАР-киназы, при этом не влияя на активность и экспрессию Akt [55].
Тиазолидиндионы оказывают влияние на распределение жировой ткани путем снижения содержания висцерального жира и увеличения подкожного [8]. Кроме того, они снижают содержание жирных кислот, связанных с СоА, в мышечной ткани, приводя к повышению чувствительности к инсулину и улучшению контроля гликемии, снижают содержание жиров в печени, эффективны в лечении неалкогольного стеатогепатита [58], нормализуют уровень трансфераз, снижают уровень воспаления в ткани печени, баллонного некроза и фиброза.
Тиазолидиндионы сохраняют функции бета-клеток у пациентов с СД 2-го типа [69]. Представитель этой группы препаратов — пиоглитазон улучшает функции бета-клеток как у пациентов, ранее не получавших лечение, так и у леченных производными сульфанилмочевины. Эффект связан с различными механизмами, включая прямые (повышение GLUT2, глюкокиназы, Pdx) с участием PPARg, и путем снижения уровня жирных кислот, связанных с СоА, в клетках. Стимуляция PPARg может обеспечивать активацию противовоспалительных механизмов при ряде воспалительных и аллергических заболеваний [4, 5].
У больных СД 2-го типа увеличено содержание липидов в миокарде, что определяет диастолическую дисфункцию. При этом в тканях миокарда определяется отчетливая инсулинорезистентность. Эта инсулинорезистентность тесно коррелирует с повышенным содержанием жиров в миокарде и не объясняется ишемической болезнью.
Заболевания венечных и каротидных сосудов представляют собой главную причину смертности пациентов с СД 2-го типа. Повышенные концентрации холестерина ЛПНП и триацилглицерола, сниженная концентрация холестерина ЛПВП определяют развитие атеросклероза у таких больных. Количественный анализ липидов атеросклеротических бляшек показывает высокое содержание НЭЖК и метаболитов жирных кислот [22], которые стимулируют воспалительные пути в развитии атеросклероза. Стимуляторы PPARg снижают уровень триацилглицерина в плазме, увеличивают концентрацию холестерина ЛПВП, превращают малые частицы ЛПНП в более плавучие и снижают уровень аполипопротеина В-100 без изменения холестерина ЛПНП [54].
Показано, что генетическая изменчивость PPARg2 имеет важную роль в развитии МС [2, 32].
Тиазолидиндионы способны также снижать уровень НЭЖК в плазме, что, возможно, будет снижать и концентрацию жирных кислот, связанных с СоА, а также других токсических метаболитов жирных кислот в гладких миоцитах артерий, ингибируя, таким образом, развитие атеросклероза. Тиазолидиндионы ингибируют высвобождение провоспалительных, прокоагулянтных, атерогенных адипоцитокинов из жировой ткани [56].
Одним из механизмов реализации противовоспалительных эффектов PPARg является способность индуцировать апоптоз моноцитов/макрофагов [1].
Таким образом, тиазолидиндионы снижают степень выраженности всех симптомов/компонентов «синдрома липотоксичности», воздействуя на механизмы, ведущие к изменению активности NF-kB.
Лечение тиазолидиндионами сопровождается некоторым увеличением массы тела (1–3 кг), которое происходит в первый год. Парадоксальным является то, что чем больше набор веса, тем больше снижение HbA1c и тем больше улучшается инсулиночувствительность и функции b-клеток [69]. Возможным объяснением этому является то, что тиазолидиндионы мобилизируют жиры из мышц, печени и b-клеток.
PPARg-рецепторы также широко представлены на клетках гипоталамических центров регуляции аппетита [30]. Таким образом, активаторы PPARg одновременно реализуют два эффекта: 1) активацию PPARg в мышцах, печени, адипоцитах, b-клетках; 2) стимуляцию аппетита.
Заключение
Инсулинорезистентность в РІ-3-киназном пути и нормальная чувствительность в МАР-киназном пути играют важную роль в развитии атеросклероза у пациентов с СД 2-го типа. Липотоксичность является главной причиной инсулинорезистентности и нарушения функций b-клеток при СД 2-го типа и имеет важное значение в развитии ССЗ. В основе всех этих на первый взгляд разнородных симптомов и патохимических феноменов лежит нарушение регуляции активности NF-kB, что приводит к «прекондиционированию системы IkB/NF-kB», определяющему развитие инсулинорезистентности, липотоксичности, системного воспаления, артериальной гипертензии. Соответственно, воздействия, приводящие к снижению транскрипционной активности NF-kB (диета, физические нагрузки, медикаментозные препараты), будут модулировать состояние прекондиционирования, устраняя молекулярную основу развития метаболического/инсулинорезистентного синдрома, снижая риск ССЗ.
1. Аторвастатин і розиглітазон індукують апоптоз моноцитів/макрофагів крові: роль поліморфізму гена PPAR-g / І.П. Кайдашев, А.М. Расін, М.В. Микитюк та ін. // Ліки. — 2007. — № 3–4. — С. 55-61.
2. Вивчення поширеності Pro12Ala поліморфізму гена PPARg2 в українській популяції з симптомами метаболічного синдрому / І.П. Кайдашев, Л.О. Куценко, О.А. Шликова та ін. // Міжнародний ендокринологічний журнал. — 2008. — 1 (13). — С. 23-26
3. Кайдашев И.П. Метаболический синдром — основы патогенеза и лечения / И.П. Кайдашев, Л. А. Куценко, И.Л. Солохина // Проблеми екології та медицини. — 2009. — № 3–4, Т. 13. — С. 23-29.
4. Кайдашев І.П. Рецептори, що активують проліферацію пероксисом, як можлива мішень у лікуванні алергійних захворювань / І.П. Кайдашев, Н.Л. Куценко // Клінічна імунологія. Алергологія. Інсектологія. — 2008. — № 3 (14). — С. 59-63.
5. Перспективы стимуляции PPARg в лечении бронхиальной астмы / И.П. Кайдашев, Н.А. Боброва, Л.В. Беркало и др. // Проблеми екології та медицини. — 2009. — Т. 13, № 1–2. — С. 32-35.
6. Полиморфизм рецептора ангиотензина ІІ 1-го типа у больных эссенциальной гипертензией в украинской популяции / И.П. Кайдашев, М.С. Расин, Л.Г. Савченко и др. // Цитология и генетика. — 2005. — 39 (5). — С. 51-55.
7. 10-year follow-up of intensive glucosae control in type 2 diabetes / R.R. Holman, S.K. Paul, M.A. Bethel et al. // N. Engl. J. Med. — 2008. — 359. — P. 1577-1589.
8. Abdominal fat distribution and peripheral and hepatic insulin resistance in type 2 diabetes mellitus / Y. Miyazaki, L. Glass, C. Triplitt et al. // Am. J. Phisiol. Endocrinol. Metab. — 2002. — 46. — E1135-1143.
9. Addition of biphasic, prandial or basal insulin to oral therapy in type 2 diabetes / R.R. Holman, K.I. Thorne, A.J. Farmer et al. // N. Engl. J. Med. — 2007. — 357. — P. 1716-1730.
10. Annual deaths attributable to obesity in the United States / D.B. Allison, K.R. Fontaine, J.E. Manson et al. // JAMA. — 1999. — 282. — P. 1530-1538.
11. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKDS 34): prospective observational study / L.M. Straton, A.I. Adler, H.A. Neil et al. // BMJ. — 2000. — 321. — P. 405-412.
12. Metabolic and molecular basis of insulin resistance / M. Bajaj, R.A. De Fronzo // J. Nucl. Cardiol. — 2003. — 10. — P. 311-323.
13. Role of adipocytes, FFA, and ectopic fat in the pathogenesis of type 2 diabetes mellitus: PPAR agonists provide a rational therapeutic approach / H. Bays, L. Mandarino, R.A. De Fronzo // J. Endocrinol. Metab. — 2004. — 89. — P. 463-478.
14. Endothelial function. From vascular biology to clinical applications / D. Behrendt, P. Ganz // Am. J. Cardiol. — 2002. — 21. — P. 40L-48L.
15. The regulation of glycogen synthase by protein phosphatase 1 in 3T3-I 1 adipocytes / M.J. Brady, A.C. Nairin, A.R. Saltiel // J. Biol. Chem. — 1997. — 272. — P. 29698-29703.
16. Cardiovascular morbidity and mortality associated with the metabolic syndrome / B. Isomaa, P. Almgren, T. Tuomi et al. // Diabetes Care. — 2001. — 24. — 683-689.
17. Characterization of selective resistance to insulin signaling in the vasculature of obese Zucker (fa/fa) rats / Z.Y. Jiang, Y.W. Lin, A. Clemont et al. // J. Clin. Invest. — 1999. — 104. — P. 447-457.
18. Connective tissue growth factor is overexpressed in complicated atherosclerotic plaques and induces mononuclear cell chemotaxis in vitro / I. Cicha, A. Yilmaz, M. Klein et al. // Art. Throm. Vas. Biol. — 2005. — Vol. 25. — P. 1008-1013.
19. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus / R.A. De Fronzo // Diabetes. — 2009. — Vol. 5 — P. 773-795.
20. Insulin resistance, lipotoxicity, type 2 diabetes and atherosclerosis: the missing links. The Claude Bernard Lecture 2009 / R.A. De Fronzo // Diabetologia. — 2010. — Vol. 53. — P. 1270-1287.
21. Is insulinresistance atherogenic? Possible mechanisms / R.A. De Fronzo // Atherosclerosis. — 2006. — Vol. 7. — P. 11-15.
22. Dietary trans fatty acids and composition of human atheromatous plaques / E. Stachowska, B. Doxegowska, D. Chlubek et al. // Eur. J. Nutr. — 2004. — Vol. 43. — P. 313-318.
23. Dose response effect of elevated plasma FFA on insulin signaling / R. Belfort, L. Mandarino, S. Kashyap et al. // Diabetes. — 2005. — Vol. 54. — P. 1640-1648.
24. Draznin B. Molecular mechanisms of insulin resistance: serine phosphorylation of insulin receptor substrate-1 and increased expression of p85 alpha: the two sides of a coin / B. Draznin // Diabetes. — 2006. — Vol. 55. — P. 2392-2397.
25. Effect of acute physiological hyperinsulinemia on gene expression in human skeletal muscle in vivo / D. Coletta, B. Balas, A.O. Chaves et al. // Am. J. Physiol. Endo. Metab. — 2008. — Vol. 294. — P. E910-E917.
26. Effect of intensive glucose lowering in type 2 diabetes / H.C. Gerstein, M.E. Miller, R.P. Byington et al. // N. Engl. J. Med. — 2008. — Vol. 358. — P. 2345-2359.
27. Effect of rosiglitazone on glucose and free fatty acid metabolism in type 2 diabetic patients / Y. Miyazaki, L. Glass, C. Triplitt et al. // Diabetologia. — 2001. — 44. — P. 2210-2219.
28. Effect of sustained reduction in plasma free fatty acid concentration on intramuscular long chain-fatty acyl-CoAs and insulin action in patients with type 2 diabetic patients / M. Bajaj, S. Suraamornkul, A. Romanelli et al. / Diabetes. — 2005. — Vol. 54. — P. 3148-3153.
29. Elevated toll-like receptor 4 expression and signaling in muscle from insulin-resistant subjects / S.M. Reyna, S. Ghost, P. Tantiwong et al. / Diabetes. — 2008. — Vol. 57. — P. 2595-2602.
30. Expression of peroxisome proliferator-activated receptor-gamma in key neuronal subsets regulating glucose metabolism and energy homeostasis / D.A. Sarruf, F. Yu, H.T. Nguyen et al. // Endocrinology. — 2008. — Vol. 150. — P. 707-712.
31. Networks biology and systems engineering: a case study in inflammation / P.T. Foteinou, E. Yang, I.P. Androulakis // Comput. Chem. Eng. — 2009. — Vol. 33 (12). — P. 2028- 2041.
32. Frequency of Pro12Ala — polymorphism of the gene PPAR?2 in the Ukrainian population and its possible relation to the development of the metabolic syndrome / I.P. Kaidashev, A.M. Rasin, O.A. Shlykova et al. // Cytology and Genetics. — 2007. — Vol. 41 (5). — P. 298-302.
33. Gard R. Insulin resistance as a proinflammatory state: mechanisms, mediators and therapeutic interventions / R. Gard, D. Tripathy, P. Dandona // Curr. Drug Targets. — 2003. — Vol. 4. — P. 487-492.
34. Glucose control and vascular complications in veterans with type 2 diabetes / W. Duckworth, C. Abraira, T. Mortiz et al. // N. Engl. J. Med. — 2009. — Vol. 360. — P. 129-139.
35. Hyperinsulinemia enhances transcriptional activity of nuclear factor–kB induced by angiotensin II, hyperglycemia, and advanced glycosylation end products in vascular smooth muscle cells / I. Golovchenko, M.L. Goalstone, P. Watson et al. // Circ. Res. — 2000. — Vol. 87. — P. 746-762.
36. Hyperinsulinemia potentiates activation of p21 Ras by growth factor / J.W. Lettner, T. Kline, K. Carel et al. // Endocrinol. — 1997. — Vol. 138. — P. 2211-2214.
37. Identification of New Rel: NF-kappa B regulatory networks by focused genome location analysis / A. De Siervi, P. De Luca, C. Moiola et al. // Cell Cycle. — 2009. — Vol. 8 (13). — P. 2093-2100.
38. Independent association of type 2 diabetes and coronary astery disease with myocardial insulin resistance / P. Lozzo, P. Chareonthaitawee, D. Dutka et al. // Diabetes. — 2002. — Vol. 51. — P. 3020-3024.
39. Insulin effect on sterol regulatory-element-binding protein-1c (SREBP-1c) transcriptional activity in rat hepatocytes / D. Azzout-marniche, D. Becard, C. Guichard et al. // Biochem. J. — 2000. — Vol. 350. — P. 389-393.
40. Insulin receptor kinase in human skeletal muscle from obese subjects with and without non-insulin dependent diabetes / J.F. Caro, M.K. Sinha, S.M. Raju et al. // J. Clin. Invest. — 1987. — 79. — P. 1330-1337.
41. Insulin-induced hexokinase II expression is reduced in obesity and NIDDM / M. Pendergrass, J. Koval, C. Vogt et al. // Diabetes. — 1998. — Vol. 47. — P. 387-394.
42. Insulin-stimulated activation of eNOS is independent of Ca2+ but requires phosphorylation by Act at Ser (1179) / M. Montagnani, H. Chen, V. A. Barr et al. // J. Biol. Chem. — 2001. — 276. — P. 30392-30398.
43. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes / A. Patel, S. MacMahon, J. Chalmers et al. // N. Engl. J. Med. — 2008. — 358. — P. 2560-2725.
44. Intensive conventional insulin therapy for type 2 diabetes. Metabolic effects during a 6-month outpatient trial / R.R. Henry, B. Gumbiner, T. Ditzler et al. // Diabetes Care. — 1993. — 16. — P. 21-31.
45. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes / D.M. Nathan, P.A. Cleary, J.Y. Backlund et al. // N. Engl. J. Med. — 2005. — 22. — P. 2643-2653.
46. The insulin resistance syndrome: physiological conside-rations / S.R. Kashyap, R.A. De Fronzo // Diabetes Vasc. Dis. Res. — 2007. — 4. — P. 13-19.
47. Plasma PAF acetylhydrolase in non-insulin dependent diabetes mellitus and obesity: effect of hyperinsulinemia and lovastatin treatment / G.B. Kudolo, P. Bressler, R.A. De Fronzo // J. Lipid Mediat. Cell Signal. — 1997. — 17. — P. 97-113.
48. Long-term inhibition of NO synthesis promotes atherosclerosis in the hypercholesterolemic rabbit thoracic aorta. PGH2 does not contribute to impaired endothelium-dependent relaxation / K. Naruse, K. Shimizu, M. Muramatsu et al. // Arterioscler. Thromb. — 1994. — 14. — 746-752.
49. Chronic insulin administration elevates blood pressure in rats / W.P. Meehan, T.A. Buchanan, W. Hsueh // Hypertension. — 1994. — 23. — P. 1012-1017.
50. Mortality and causes of death in the WHO multinational study of vascular diseases in diabetes / N.J. Morrish, S.L. Wang, L.K. Stevens et al. // Diabetologia. — 2001. — 44 (Suppl. 2). — S14-S21.
51. Mutation of insulin receptor at tyrosine 960 inhibits signal transmission but does not effect its tyrosine kinase activity / M.F. White, J.N. Livingston, G.M. Backer et al. // Cell. — 1992. — 54. — P. 641-649.
52. Nuclear factor KappaB signaling in atherogenesis / M.P. de Winter, E. Kanters, G. Kraal et al. // Art. Throm. Vasc. Biol. — 2005. — 25. — P. 904-914.
53. Physiological hyperinsulinemia impairs insulin-stimulated glucogen syntase activity and glycogen synthesis / P. Lozzo, T. Pratipanawatr, H. Pijl et al. // Am. J. Physiol. — 2001. — 280. — P. E712-E719.
54. Pioglitazone and rosiglitazone have different effects on serum lipoprotein particle concentrations and sizes in patients with type 2 diabetes and dyslipidemia / M.A. Deeg, J.B. Buse, R.B. Goldberg et al. // Diabetes Care. — 2007. — 30. — P. 2458-2464.
55. PPAR-gamma agonists inhibit toll-like receptor mediated activation of dendritic cells via the MAP kinase and NF-kappaB pathways / S. Appel, V. Mirakaj, A. Bringmann et al. // Blood. — 2005. — 106 (12). — P. 3888-3894.
56. Predictors of improved glycaemic control with rosiglitazone therapy in type 2 diabetic patients: a practical approach for the primary care physician / M. Miyazaki, E. de Filippis, M. Bajaj et al. // Br. J. Diabetes Vasc. Dis. — 2005. — 5. — P. 28-35.
57. Quantitation of muscle glycogen synthesis in normal subjects with non-insulin-dependent diabetes by 13C nuclear magnetic resonance spectroscopy / J.I. Shulman, D.I. Rothman, T. Jue et al. // N. Engl. J. Med. — 1990. — 322. — P. 223-238.
58. Randomized, placebo-controlled trial of pioglitazone in nondiabetic subjects with nonalcoholic steatohepatitis / G.P. Aithal, J.A. Thomas, P.V. Kaye et al. // Gastroenterology. — 2008. — 135. — P. 1176-1184.
59. Recurrence of cardiovascular events in patients with type 2 diabetes / C.B. Giorda, A. Аvogaro, M. Maggini et al. // Diabetes Care. — 2008. — Vol. 31. — P. 2154-2159.
60. Relation between insulin resistance and carotid intima-media thickness and stenosis in non-diabetic subjects. Results from a cross-sectional study in Malmo, Sweden / B. Bedblad, P. Nilsson, L. Janzon et al. // Diabet Med. — 2002. — 17. — P. 299-307.
61. Reversal of obesity — and diet-induced insulin resistance with salicylates or targeted disruption of Ikk beta / M. Ynan, N. Konstantopoulos, J. Lee et al. // Science. — 2001. — 293. — P. 1673-1677.
62. Reversibility of defective adipocytes insulin receptor kinase activity in non-insulin dependent diabetes mellitus. Effect of weight loss / G.R. Freidenberg, D. Reichart, J.M. Olefsky et. al. // J. Clin. Invest. — 1988. — 82. — P. 1398-1406.
63. Rosiglitazone improves myocardial glucose uptake in patients with type 2 diabetes and coronary artery disease / R. Lautamaki, K.E. Juhani Airaksinen, M. Seppanen et al. // Diabetes. — 2005. — 54. — P. 2787-2794.
64. Serine phosphorylation of insulin receptor substrate 1 by inhibitor kappaB kinase complex / Z. Gao, D. Hwang, F. Bataille et al. // J. Biol. Chem. — 2002. — 277. — P. 48115-48121.
65. Suppression of nuclear factor-kappa b by troglitazone: evidence for an anti-inflammatory effect and a potential antiatherosclerotic effect in the obese / H. Ghanim, R. Garg, A. Aljada et al. // J. Clin. Endocrinol. Metab. — 2001. — 86. — P. 1306-1312.
66. Critical nodes in signaling pathways: insights into insulin action / C.M. Taniguchi, B. Emanuelli, C.R. Kahn // Nat. Rev. Mol. Cel. Boil. — 2006. — 7. — P. 85-96.
67. The influence of pioglitazone on NF-kB expression in CD40+ lymphocytes is polymorphism-dependent / I.P. Kaidashev, N. Kutzenko, O.A. Shlykova et al. // J. Allergy Clin. Immunol. — 2009. — 123. — № 2. — S. 92.
68. The metabolic profile of NIDDM is fully established in glucose-tolerant offspring of two Mexican-American NIDDM parents / G. Gulli, F. Ferrannini, M. Stern et al. // Diabetes. — 1992. — 41. — P. 1575-1586.
69. Thiazolidinediones improve beta-cell function in type 2 diabetic patients / A. Gastaldelli, E. Ferrannini, Y. Miyazaki et al. // Am. J. Phisiol. Endocrinol. Metab. — 2007. — 292. — P. 871-883.
70. Triglyceride-rich lipoprotein lipolysis increases aggregation of endothelial membrane microdomains and produces reactive oxygen species / L. Wang, A.R. Sapuri-Butin, H.H. Aung et al. // Am. J. Physiol. Heart Circ. Physiol. — 2008. — 295. — P. H237-H244.
71. Triplitt C. Exenatide: first in class incretin mimetic for the treatment of type 2 diabetes mellitus / C. Triplitt, R.A. De Fronzo // Expert. Rev. Endocrinol. Metab. 2006. — 1. — P. 329-341.
72. Tumor necrosis factor alpha produces insulin resistance in skeletal muscles by activation of inhibitor kappa B kinase in a p38 MAPK-dependent manner / C. De Alvaro, T. Teruel, R. Hernandez et al. // J. Biol. Chem. — 2004. — 279. — P. 17070-17078.
73. Lipid overload and overflow: metabolic trauma and the metabolic syndrome / R.H. Under // Trends Endocrinol Metab. — 2003. — 14. — P. 398-403.
74. Obesity-induced inflammatory changes in adipose tissue / K.E. Wellen, G.S. Hotamisligil // J. Clin. Invest. — 2003. — 112. — P. 1785-1788.
75. Ikappa B kinases: key regulators of the NF-kappaB pathway / Y. Yamamoto, R.B. Gaynor // Trends Biochem. Sci. — 2004. — 29. — P. 72-79.