
Журнал «Медицина неотложных состояний» 4(35) 2011
Вернуться к номеру
Патогенетические особенности развития циркуляторной гипоксии мозга при артериальной гипертензии
Авторы: Головченко Ю.И., Трещинская М.А. Национальная медицинская академия последипломного образования им. П.Л. Шупика, г. Киев
Рубрики: Семейная медицина/Терапия, Медицина неотложных состояний
Версия для печати
В статье представлен анализ данных современной литературы относительно влияния повышенного артериального давления (АД) на головной мозг и его гемодинамику. При артериальной гипертензии (АГ) происходит ремоделирование сосудистого дерева, нарушение ауторегуляции мозгового кровотока и поражение вещества головного мозга. Не следует упускать из виду вероятность развития АГ вследствие нарушения функционирования сосудодвигательного центра и надсегментарных структур автономной нервной системы в результате первичного повреждения головного мозга.
Артериальная гипертензия, головной мозг, церебральная гемодинамика.
Потребность органов и тканей в кислороде и питательных веществах обеспечивается посредством адекватной перфузии благодаря поддержанию постоянного уровня артериального давления (АД) и непрерывного перераспределения артериальной крови между всеми органами и тканями в соответствии с их потребностями. Адекватное кровоснабжение органов и тканей реализуется сердечно-сосудистой системой, каждый участок которой характеризуется различными скоростными и объемными свойствами кровотока, определенным систолическим и диастолическим давлением и сосудистым сопротивлением (рис. 1) [16].
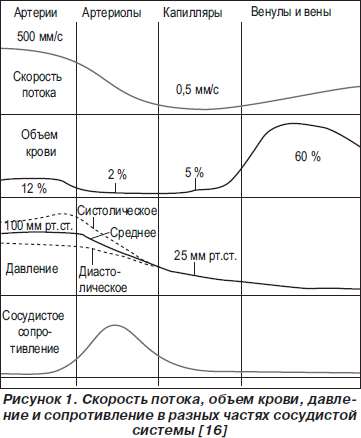
Кровоток через любой орган в значительной степени определяется его сосудистым сопротивлением, которое главным образом зависит от диаметра артериол. Соответственно, органный кровоток регулируется факторами, влияющими на тонус гладкой мускулатуры артериол.
Механизмы, регулирующие кровообращение, можно разделить на две категории: 1) центральные, определяющие величину артериального давления и системное кровообращение; 2) локальные, контролирующие величину кровотока через отдельные органы и ткани [16, 28].
К центральным механизмам регуляции кровотока относятся неврогенный и гуморальный, к локальным — метаболический, миогенный и эндотелиальный механизмы.
Активацию центральных механизмов регуляции вызывают повышение или снижение АД, общие изменения газового состава крови (гипоксия, гипероксия, гиперкапния), а также различные воздействия на организм человека физических и эмоциональных факторов (изменение температуры окружающей среды, болевое воздействие, страх, стресс и т.п.).
Изменение системного АД приводит к активации барорецепторов, расположенных в синокаротидной зоне и дуге аорты. Сами рецепторы представляют собой механорецепторы, реагирующие на изменение артериального давления по степени растяжения эластичных стенок артерий [16].
Импульсы, генерируемые барорецепторами каротидного синуса, проходят по нервам Геринга, которые затем присоединяются к языкоглоточному нерву перед входом в центральную нервную систему. Афферентные волокна от аортальных барорецепторов идут в центральную нервную систему в составе блуждающих нервов. Центральные регулирующие структуры находятся в продолговатом мозге — так называемом сосудодвигательном центре, а также в высшем корковом центре вегетативной регуляции — гипоталамусе. Именно там проходят процессы, которые фактически приводят к интеграции сенсорной информации и превращению ее в соответствующие симпатические и парасимпатические реакции. Результирующая сосудистая реакция на повышение системного АД характеризуется снижением тонуса симпатической нервной системы, на снижение АД — повышением тонуса симпатического отдела автономной нервной системы с развитием соответствующей по направленности сосудистой реакции — вазоконстрикции или вазодилатации. В их формировании принимают участие не только центры вегетативной нервной системы, но и рецепторы, проводящие пути и центральные чувствительные анализаторы, воспринимающие первичную информацию и передающие ее в гипоталамус, где и формируется вторичный сосудистый ответ [16].
Сосудистые реакции как результат неврогенного механизма регуляции кровообращения, развивающиеся при общих воздействиях на организм, имеют адаптивный характер. При длительном воздействии на указанные рецепторы — например, постоянном повышении системного АД в результате истощения или адаптации, артериальный барорецепторный рефлекс не может выполнять роль механизма долговременной регуляции величины АД.
Гуморальная регуляция сосудистого тонуса осуществляется путем выделения в кровь вазоактивных веществ, обладающих сосудосуживающим или сосудорасширяющим свойством. К сосудосуживающим веществам относятся гормоны мозгового вещества надпочечников — адреналин и норадреналин, а также задней доли гипофиза — вазопрессин. Адреналин и норадреналин сужают артерии и артериолы кожи, органов брюшной полости и легких, а вазопрессин действует преимущественно на артериолы и капилляры. Кроме того, гуморальным сосудосуживающим фактором являются серотонин, продуцируемый в слизистой оболочке кишечника и некоторых участках головного мозга, и ренин, продуцируемый в почках. К сосудорасширяющим веществам относятся медулин, ряд простагландинов, брадикинин, ацетилхолин, гистамин и ряд других веществ [19, 28].
Важное значение имеют локальные механизмы регуляции тонуса различных сегментов артериального сосудистого русла. Все отделы артериальной сосудистой системы в покое обладают определенным базисным тонусом, являющимся собственным свойством гладкомышечных клеток сосудистой стенки. При изменении локального метаболизма или величины внутрипросветного давления активируются локальные механизмы компенсации, приводящие к локальным изменениям тонуса сосудов. Артериолы, регулирующие кровоток через данный орган, сами находятся в тканях этого органа. Таким образом, гладкая мускулатура артериол подвергается влиянию химического состава интерстициальной жидкости органа, который они снабжают. Концентрация различных веществ в интерстициальной жидкости отражает баланс между метаболической активностью ткани и ее кровоснабжением [16].
Практически во всех сосудистых зонах недостаток кислорода снижает тонус артериол и вызывает расширение сосудов, в то время как высокое содержание О2 влечет за собой их сужение. Многие вещества, помимо кислорода, присутствующие в тканях, могут воздействовать на тонус гладкой мускулатуры сосудистой стенки. Например, увеличение скорости метаболизма в скелетной мышце при физической нагрузке приводит не только к снижению уровня кислорода в ткани, но и к повышению содержания СО2, Н+ и К. Во время физической нагрузки возрастает и осмолярность мышечной ткани. Все эти изменения химического состава крови приводят к расширению артериол. Кроме того, при повышении метаболической активности или снижении содержания кислорода из клеток многих тканей может высвобождаться аденозин — чрезвычайно активный сосудорасширяющий агент [19].
Важным локальным регуляторным звеном являются эндотелиальные клетки, которые покрывают всю внутреннюю поверхность сердечно-сосудистой системы. Эндотелиальные клетки продуцируют ряд факторов, влияющих на гладкомышечные клетки сосудистой стенки и вызывающих развитие разнонаправленных сосудистых реакций. Снижение тонуса гладкомышечных клеток в основном вызывает эндотелиальный фактор релаксации — оксид азота (NO). Он образуется в эндотелиальных клетках из L-аргинина под действием эндотелиальной NO-синтазы, которая активируется при увеличении концентрации внутриклеточного Са2+.
Ацетилхолин и некоторые другие вещества (включая брадикинин, вазоактивный кишечный пептид, субстанцию Р) стимулируют продукцию NO через рецепторы, управляющие кальциевыми каналами на эндотелиоцитах. Одним из основных индукторов выработки NO является напряжение сдвига, возникающее при изменении уровня кровотока за счет активации чувствительных к растяжению кальциевых каналов. В физиологических условиях подобные реакции могут возникать при физической нагрузке [28].
Факторы, которые блокируют продукцию NO посредством торможения NO-синтазы, вызывают существенное увеличение сосудистого сопротивления в большинстве органов в состоянии покоя. Поэтому считается, что эндотелиальные клетки в норме всегда вырабатывают определенное количество NO, а это в сочетании с другими факторами имеет важное значение в обеспечении нормального результирующего тонуса артерий и артериол в организме [19]. Одним из наиболее мощных вазоконстрикторов, синтезируемых эндотелием, является эндотелин.
Еще одним способом локальной регуляции сосудистого тонуса является миогенный механизм. В 1902 г. W. Bayliss [35] сообщил о результатах эксперимента, который показал, что «мышечный слой артерий, как и другие гладкие мышцы, отвечает на растяжение сокращением» независимо от иннервации. Позднее В. Folkow [46] продемонстрировал сужение денервированного сосуда в ответ на повышение внутрипросветного давления. Выявленная реакция была названа эффектом Остроумова — Бейлисса. В соответствии с этим эффектом в ответ на снижение внутрипросветного давления происходит расслабление гладкомышечных клеток сосудистой стенки и дилатация просвета сосуда, повышение давления вызывает вазоконстрикцию. Механизм миогенной реакции недостаточно ясен, но чувствительные к растяжению ионные каналы в клетках гладкой мускулатуры артериол представляются наиболее вероятными участниками процесса.
Современные представления о регуляции мозгового кровообращения основываются на признании его автономии, наличии многозвеньевой системы регуляции, включая внечерепной уровень, а также системы ауторегуляции, действующей в определенных пределах, и множественности механизмов ее реализации. У здорового человека значения среднего системного АД, в пределах которых действуют механизмы ауторегуляции мозгового кровотока, составляют 50–70 и 150–170 мм рт.ст. соответственно при нормальном рН крови — в диапазоне от 7,35 до 7,45 [8, 17, 36, 55].
Верхний предел ауторегуляции мозгового кровотока определяется как уровень среднего гемодинамического АД, при превышении которого скорость мозгового кровотока начинает возрастать. У лиц с нормальным АД при повышении среднего АД выше 150–170 мм рт.ст. высокое перфузионное давление может преодолеть сопротивление мозговых артерий. Возникает так называемая силовая дилатация артерий, которая сопровождается резким возрастанием мозгового кровотока, отеком головного мозга и нарушением целостности гематоэнцефалического барьера (ГЭБ). В результате отека мозговой кровоток может вторично уменьшаться [5, 8, 18, 20].
В клинических исследованиях показано, что у человека критическая скорость мозгового кровотока, при которой появляется неврологическая симптоматика, составляет для серого вещества 15–29 мл/мин, т.е. примерно 30–40 % от нормы. У здорового человека при снижении среднего АД ниже 50–70 мм рт.ст. вазодилатация мозговых артерий оказывается недостаточной, чтобы поддерживать необходимую для удовлетворения метаболических потребностей головного мозга скорость мозгового кровотока. При этом доставка кислорода к головному мозгу может поддерживаться на достаточном уровне благодаря увеличению экстракции кислорода из крови тканью мозга. Однако если среднее АД опускается ниже 30–40 мм рт.ст., повышение экстракции кислорода из крови тканью головного мозга перестает компенсировать снижение мозгового кровотока. В результате этого развивается кислородное голодание головного мозга, которое проявляется симптомами и признаками церебральной ишемии в виде гипервентиляции, головокружения, потери сознания и т.д.
При АГ кривая ауторегуляции сдвигается вправо, так как сосуды головного мозга адаптируются к более высоким уровням системного АД. Этот феномен получил название «адаптация ауторегуляции мозгового кровотока». У больных с нелеченной или плохо леченной АГ нижний предел ауторегуляции составляет 113,17 мм рт.ст., что достоверно выше, чем у больных без АГ [8, 21]. Смещение нижнего предела ауторегуляции вправо означает, что у лиц с АГ острая ишемия головного мозга возникает при более высоких значениях среднего системного АД. Клинически это проявляется тем, что у больных АГ признаки гипоперфузии головного мозга возникают при быстром снижении системного АД до такого уровня, который легко переносится больными без АГ. Так, симптомы церебральной ишемии у больных АГ появляются при снижении среднего системного АД до 65,10 мм рт.ст., а у больных без АГ — до 43,8 мм рт.ст. [2, 70, 71]. У больных АГ, получающих эффективную антигипертензивную терапию, нижний предел ауторегуляции значительно ниже, чем у плохо леченных больных, и составляет в среднем 96,17 мм рт.ст. [21]. Смещение нижнего предела ауторегуляции мозгового кровотока у больных АГ следует учитывать при лечении гипертонических кризов: чтобы избежать развития ишемии головного мозга, рекомендуется снижать высокое АД не более чем на 15–20 % от исходного уровня за 2–3 ч.
В обеспечении регуляторных реакций принимают участие все сегменты церебральной артериальной системы, начиная от магистральных артерий головы и заканчивая артериолярным руслом. Развивающиеся вазодилататорные или вазоконстрикторные реакции являются результатом сочетанного действия миогенного, неврогенного и метаболического механизмов ауторегуляции [16].
Регулирующая функция магистральных артерий головы не исчерпывается поддержанием оптимального уровня системного АД. Они активно участвуют в регуляции притока крови к виллизиеву кругу и другим сосудам мозга путем изменения своего просвета. Анатомической основой механизма вазоконстрикторных реакций являются мощный мышечный слой и богатая иннервация, что характерно для прекраниальных отрезков сонных и позвоночных артерий. Сужение магистральных артерий головы происходит в ответ на повышение АД, а также при венозном застое и отеке головного мозга. Физиологический смысл этой реакции заключается в ограничении притока крови в сосудистую систему мозга. Расширение магистральных артерий возникает при падении АД. Регуляция притока крови к мозгу обеспечивается также участками магистральных артерий головы, расположенными в кавернозном и атлантозатылочном синусах. Повышение венозного давления в синусах служит сигналом к ограничению притока крови к мозгу в указанных участках сонных и позвоночных артерий. Важную роль в обеспечении равномерности кровотока играют физиологические изгибы магистральных артерий, которые ограничивают пульсовые и другие колебания АД [7].
У больных АГ по сравнению с лицами с нормальным АД вероятность развития инсульта больше в 3–7 раз, в 6 раз чаще возникает сердечная недостаточность, в 4 раза выше риск развития ишемической болезни сердца (ИБС), в 2 раза — поражение периферических артерий [27, 30, 50]. Риск возникновения основных сердечно-сосудистых осложнений увеличивается приблизительно на 30–40 % каждые 10 мм рт.ст. повышенного систолического артериального давления (САД) у больных разных возрастных групп и полов. При стойком повышении диастолического артериального давления (ДАД) на 5 мм рт.ст. риск мозгового инсульта увеличивается на 34 %, а инфаркта миокарда — на 21 %. При повышении ДАД на 10 мм рт.ст. риск возрастает соответственно до 56 и 37 %. Уровень ДАД, превышающий 80 мм рт.ст., достоверно связан с развитием 57 % случаев смерти от мозгового инсульта и около 24 % случаев смерти от ИБС. Существует достоверная позитивная корреляция между уровнем АД и общей смертностью: риск неуклонно возрастает по мере повышения АД [24].
Повышенное АД приводит к ускоренному развитию цереброваскулярных заболеваний и, как следствие — к формированию хронической ишемии головного мозга (по МКБ-10, рубрика 167.8) или острых нарушений мозгового кровообращения (по МКБ-10, рубрика 160) [30]. В то же время особую значимость имеет прямое действие АГ на молекулярные, биохимические и клеточные механизмы функционирования головного мозга, на микроциркуляторно-тканевые взаимоотношения.
Установлено, что риск развития мозгового инсульта имеет линейную логарифмическую зависимость от уровней ДАД и САД [54, 56]. Достоверных различий в частоте выявления АГ при разных типах мозгового инсульта выявлено не было: она составляет при ишемическом инсульте 92,3 %, при геморрагическом — 92,6 %.
Проведение антигипертензивной терапии в рамках популяционной стратегии (снижение ДАД во всей популяции на 2 %) или целевой стратегии высокого риска (снижение ДАД на 7 % у лиц с исходным его уровнем выше 95 мм рт.ст.) позволяет достоверно предотвратить 1 из 6 смертей от мозгового инсульта и 1 из 20 смертей от ИБС [76].
Выраженное повреждающее действие оказывают предутренний подъем АД, высокие суточные градиенты АД и отсутствие ночного снижения АД, что во многом связывается с колебаниями сосудистого тонуса [5, 17, 23, 32, 36]. Связь между АД и опасностью развития цереброваскулярных нарушений может основываться не только на уровневом эффекте АД, но и на продолжительности течения АГ, другими словами, стаже заболевания [21, 22, 58]. Подтверждением сказанного являются данные о том, что большинство случаев инсульта были зарегистрированы у больных с пограничной или мягкой АГ. При этом польза от снижения АД отмечена у лиц как с повышенным, так и нормальным АД. Поэтому, несмотря на то, что более высокий уровень АД предполагает больший относительный риск развития инсульта, современные данные склоняют в пользу концепции о том, что длительное течение АГ для развития инсульта играет большую роль, чем одномоментное повышение АД [21].
Результаты исследований показывают, что медиаторы АГ, такие как ангиотензин II, могут влиять на риск заболевания мозговым инсультом независимо от уровня повышения АД. АГ и ее медиаторы оказывают значительное негативное влияние на молекулярные и клеточные процессы в ткани головного мозга, что приводит к развитию хронической церебральной ишемии [2, 48, 51]. Таким образом, АГ является важным регулируемым фактором риска развития цереброваскулярной патологии, оказывая значительное негативное действие как на мозговое кровообращение, так и на молекулярные и клеточные процессы в ткани головного мозга.
Можно выделить следующие основные формы влияния АГ на церебральные сосуды: 1) формирование гипертонической микроангиопатии — Small artery brain disease [10, 11]; 2) аневризматизация (истончение) сосудистой стенки; 3) усугубление атеротромботического поражения артерий крупного калибра — Large artery brain disease, дестабилизация атеросклеротических бляшек и формирование тромбоэмболов [11, 40, 44, 72]. Часто перечисленные патологические изменения развиваются параллельно. Таким образом, разделение гипертонических и атеросклеротических цереброваскулярных нарушений всегда достаточно условно, в большинстве случаев речь идет о смешанных формах дисциркуляторной энцефалопатии [3, 9, 10]. К нарушениям церебрального кровообращения, для которых АГ является непосредственной причиной, относят кровоизлияния, развивающиеся вследствие разрыва измененной сосудистой стенки (спонтанные внутримозговые гематомы и субарахноидальные кровоизлияния), а также некоторые варианты инфаркта мозга (прежде всего микроциркуляторные лакунарные инфаркты) [1, 5, 72]. Наряду с этим АГ приводит к развитию хронической церебральной ишемии, проявляющейся симптомами хронической гипертензивной энцефалопатии и сосудистой деменции.
К первичным деструктивным изменениям в церебральных сосудах при АГ относят плазматическое пропитывание сосудистой стенки [6]. При этом может наблюдаться набухание и гомогенизация одного субэндотелиального слоя, его утолщение. В более тяжелых случаях плазморрагии сопровождаются проникновением в стенку сосудов липидов и эритроцитов, фибриноидным некрозом стенки, что может приводить к образованию милиарных и расслаивающих аневризм. В подобные изменения иногда вовлекаются все оболочки сосуда [6, 12]. Другими видами острых деструктивных процессов при АГ являются изолированный некроз и дистрофические изменения миоцитов в средней оболочке артерий, что может привести к гибели этой оболочки. При этом характерно развитие изменений в средней оболочке артерий при отсутствии признаков плазматического пропитывания оболочки и сохранности внутренней эластической мембраны.
Результатом массивных плазморрагий, приводящих к набуханию стенки с резким сужением или облитерацией просвета сосуда, является гипертонический стеноз. В бассейне стенозированного или облитерированного сосуда резко снижается кровоток, что приводит к развитию очаговой ишемии мозга и формированию малых глубинных (лакунарных) инфарктов или зон неполного некроза ткани мозга [6].
Дегенерация стенки артериол с отложением гиалина под базальной мембраной и в межклеточном веществе средней оболочки является проявлением необратимых системных изменений при АГ и может считаться признаком гиалинового артериолосклероза [29].
В качестве возможных пусковых реакций, обусловливающих изменение сосудистой проницаемости, можно отметить нарушение регуляции метаболизма внутриклеточного кальция. Показано уменьшение активности Са2+-АТФазы на внешней мембране эндотелия и гладкомышечных клеток артериол, сочетающееся с повышением проницаемости артериол [60–63].
Важное значение имеют зависимые от эндотелия сосудосуживающие реакции, вовлечение гормонального звена с активацией адренергической и ренин-ангиотензиновой систем [6].
АГ изменяет способность эндотелиальных клеток высвобождать вазоактивные факторы и повышает сократительный тонус системных и церебральных артерий [43]. АГ также изменяет сосудисто-мозговую ауторегуляцию, которая является особенностью артериол головного мозга, позволяющей поддерживать мозговой кровоток и перфузионное давление в относительном постоянстве, несмотря на колебания системного АД в определенных пределах [56]. Хроническая АГ смещает эти пределы в сторону более высокого давления, делая головной мозг более уязвимым к понижению перфузионного давления [37]. Эти структурные и функциональные изменения ведут к повышению чувствительности головного мозга к повреждениям ишемического характера. Например, в результате смещения пределов ауторегуляции и повышения эластичности сосудов падение перфузионного относительно ДАД в закупоренной артерии, скорее всего, приведет к значимому ухудшению мозгового кровотока.
Повреждение нервного аппарата клетки происходит рано и в большей степени, чем других структур сосудистой стенки. В результате денервации сосуда развиваются соответствующие функциональные нарушения, в частности повышение чувствительности к катехоламинам [6]. Разрыв стенки денервированных сосудов может стать источником массивного кровоизлияния в мозг.
При АГ в мозговых артериях наряду с функциональными нарушениями происходят структурные изменения, которые названы ремоделированием. Под термином «ремоделирование», пришедшим на смену понятиям гипертрофии левого желудочка и гипертрофии сосудистой стенки, подразумевается комплекс деструктивных, адаптивных и репаративных реакций, вовлекающих сосуды любого диаметра — как крупные экстра- и интракраниальные, так и более мелкие (до 500 мкм в диаметре) артерии и сосуды микроциркуляторного русла. Ремоделирование сердечно-сосудистой системы является неизменным атрибутом гипертонической болезни, являясь, с одной стороны, осложнением АГ, а с другой — фактором ее прогрессирования, важным механизмом, ответственным за изменение сосудистого резерва и ауторегуляции церебрального кровотока, развитие атеросклероза [13, 21]. Влияя на отдельные звенья ремоделирования, возможно предотвратить многие осложнения атеросклероза, снизить риск развития цереброваскулярных заболеваний [1, 4, 5, 14].
По данным электронной и световой микроскопии, морфологическими признаками ремоделирования церебральных сосудов являются уменьшение их просвета, увеличение мышечного слоя, уменьшение ядер эндотелиальных клеток и наличие адвентициальноподобных клеток в медиальном слое сосудистой стенки [2, 47]. В настоящее время расшифрован ряд механизмов развития процессов ремоделирования — адренергическая стимуляция гипертрофии и пролиферации гладкомышечных клеток сосудов под влиянием тромбоцитарного ростового фактора, изменение активности матричных металлопротеиназ, влияние урокиназного активатора плазминогена [1].
Пролиферативные процессы влияют на эластичность и растяжимость сосудов, усугубляют проявления атеросклероза [45]. Изменения в стенке магистральных сосудов головы при АГ включают развитие дисфункции эндотелия, утолщение комплекса интима-медиа (КИМ) прежде всего сонных артерий и, как следствие, прогрессирование атеросклероза и формирование атеротромбоза.
Выявлена связь между типом ишемического мозгового инсульта, его локализацией, размером очага ишемического повреждения и индексом интима-медиа в сонных артериях [41, 65, 74].
Истончение стенки сосуда в результате изменения структуры базальной мембраны и коллагенового матрикса ведет к появлению микроаневризм и может являться причиной развития геморрагического мозгового инсульта [64]. В настоящее время данные о корреляционной связи между КИМ и риском развития геморрагического мозгового инсульта недостаточны; требуется проведение дополнительных исследований [75].
Длительное существование АГ включает основные механизмы развития хронического патологического (нейродегенеративного) процесса в ткани головного мозга: хроническое воспаление, изменение проницаемости гематоэнцефалического барьера и аутоиммунизацию организма к собственным нейроспецифическим белкам с последующим вторичным аутоиммунным повреждением головного мозга, митохондриальную дисфункцию и оксидантный стресс, программированную клеточную смерть (апоптоз) и дефицит трофических факторов.
Известно, что у организма отсутствует иммунологическая толерантность к ткани головного мозга, поскольку гематоэнцефалический барьер (ГЭБ) защищает его от иммунного конфликта. Головной мозг может быть распознан иммунной системой организма как чужеродный объект в случае нарушения целостности ГЭБ за счет эндотелиальной и/или астроцитарной дисфункции. Особенностями структуры ГЭБ являются наличие плотных контактов между эндотелиальными клетками, отсутствие фенестрированности, низкий уровень везикулярного транспорта через эндотелий капилляров. В патологических условиях проницаемость ГЭБ может повышаться по механизмам нарушения плотных контактов между эндотелиальными клетками или повышения активного транспорта через эндотелий [42]. Было показано, что нарушение ГЭБ является необходимым условием для реализации воспалительного процесса [52].
Проницаемость ГЭБ изменяется на фоне АГ. Одним из наиболее важных факторов в нарушении проницаемости ГЭБ при АГ и связанных с ней воспалительных реакций считается изменение экспрессии молекул клеточной адгезии, расположенных на эндотелии, — ICAM-1, ICAM-2, VCAM-1.
Доминирование воспалительно-аутоиммунного компонента приводит к диффузному поражению перивентрикулярного белого вещества мозга с характерными признаками на МРТ головного мозга в виде зон лейкоарейоза, преимущественно лобной локализации. Такое повреждение влечет за собой функциональный разрыв префронтальных субкортикальных связей, играющих существенную роль в осуществлении психомоторных функций [66]. Недавно показано, что изменения в таламусе, преимущественно в его вентромедиальных ядрах, имеют наибольшую значимость в развитии у больных когнитивных расстройств [34].
Таким образом, у больных с хронической ишемией головного мозга на фоне АГ развивается генерализованная аутоиммунизация к структурным компонентам нервной ткани. Аутоиммунные процессы принимают участие в формировании фонового сосудистого повреждения головного мозга (энцефалопатии) и подготавливают церебральную ткань к развитию крупноочагового инфаркта в случае острого снижения мозгового кровотока [25].
Исследования A. Siren и соавт. [69] показали, что длительная АГ может вызывать активацию внутриклеточных молекул адгезии (intercellular adhesion molecule ICAM-1) на эндотелиальных клетках и периваскулярное накопление лейкоцитов в ткани мозга, что отражает своеобразную предрасположенность к нарушению микроциркуляции у больных с АГ.
Установлено, что при нарушениях микроциркуляции и ишемизации ткани головного мозга микроглия начинает продуцировать широкий спектр токсичных для ткани мозга соединений: провоспалительные цитокины, лиганды для глутаматного NMDA рецепторного комплекса, протеазы, катепсин В, лизозимы, эйкозаноиды (в том числе тромбоксан В2), супероксидный анион, нитроксид и инициирует цитотоксическое действие астроцитов [33, 39, 49]. Анализ перечня соединений, синтезируемых микроглиальными клетками, свидетельствует об активном и согласованном с другими клеточными пулами участии активированной микроглии во всех основных процессах глутамат-кальциевого каскада, поддерживающем глутаматную эксайтотоксичность, активацию внутриклеточных ферментов, свободнорадикальные реакции, перекисное окисление липидов. Однако наряду с этим микроглия выполняет и специализированные иммунные функции, индуцируя и поддерживая воспалительную реакцию в ткани головного мозга, что в конечном итоге ведет к отсроченным нейрональным потерям, изменениям микроциркуляции и гематоэнцефалического барьера [59].
Рассматривая взаимоотношения АГ и морфофункционального состояния головного мозга, следует выделить два аспекта: влияние АГ на состояние головного мозга и участие головного мозга в формировании АГ [1].
В последние годы пристальное внимание исследователей направлено на изучение нейрогенных механизмов АГ. Известна роль высших вегетативных центров, локализованных в лимбико-ретикулярном комплексе, и симпатического отдела автономной нервной системы в развитии АГ. Однако ранее изучались преимущественно периферические механизмы влияния симпатической нервной системы на сократительную способность сосудов и миокарда, опосредуемые через систему аминергической нейротрансмиссии (катехоламины) и адренергические рецепторы. Расположение катехоламинергических нейронов в головном мозге в настоящее время уточняется.
Норадренергические нервные клетки находятся только в узкой переднелатеральной зоне покрышки продолговатого мозга и моста (А1 — А7). Самая большая группа норадренергических клеток А6 расположена в области голубого пятна (ядра) и включает почти половину всех норадренергических клеток (около 1000). У взрослых голубое пятно включает нейромеланинсодержащие нервные клетки, формирующие темно-синюю полоску длиной около 1 см в верхней части моста у дна четвертого желудочка. Ядро простирается вверх, вплоть до нижних холмиков четверохолмия. Аксоны клеток голубого пятна многократно ветвятся, их адренергические окончания можно найти во многих отделах центральной нервной системы. Волокна, идущие от этих групп норадренергических нервных клеток, или поднимаются к среднему мозгу, или нисходят к спинному мозгу. Кроме того, норадренергические клетки имеют связи с мозжечком. Норадренергические нейроны имеют более обширные связи, чем дофаминергические. Близость норадренергических волокон к артериолам и капиллярам мозга объясняет их важную роль в регуляции мозгового кровотока. Считается, что нейроны голубого пятна принимают активное участие в процессах формирования памяти и обеспечении когнитивных функций, т.е. регулируют такие функции, как мышление, интегрирование, внимание, планирование, обучаемость, поведенческие реакции [53].
Адреналин (эпинефрин)-синтезирующие нейроны находятся только в продолговатом мозге, в узкой переднелатеральной области. Наибольшая группа клеток С1 лежит позади заднего оливного ядра, средняя группа клеток С2 — рядом с ядром одиночного пути, и группа клеток С3 — непосредственно под перивентрикулярным серым веществом. Эфферентные пути от С1 — С3 идут к заднему (дорсальному) ядру блуждающего нерва, ядру одиночного пути, голубому пятну, перивентрикулярному серому веществу моста и среднего мозга, гипоталамусу и паравентрикулярным ядрам. Физиологические эксперименты показали, что группа клеток С1 является очень чувствительным вазопрессорным центром.
Для катехоламинов описано 4 главных типа рецепторов: a1, a2, b1 и b2. Они различаются по реакции на различные агонисты или антагонисты и по постсинаптическим эффектам. Рецепторы a1 управляют кальциевыми каналами при помощи вторичного мессенджера инозитол-1,4,5-трифосфата (IP3) и при активации повышают внутриклеточную концентрацию ионов Ca2+. Рецепторы a2 ведут к уменьшению концентрации вторичного мессенджера цАМФ, что вызывает различные эффекты. Рецепторы b при помощи вторичного мессенджера цАМФ повышают проводимость мембран для ионов К+, генерируя тормозной постсинаптический потенциал.
Доказательства того, что стимуляция a2-адренергических рецепторов в гипоталамусе и области nucleus tractus solitarii (NTS) продолговатого мозга оказывает ингибирующий эффект на эфферентную симпатическую активность и приводит к вазодилатации, позволили предположить, что повышение АД может являться следствием пониженной чувствительности нейронов этих областей головного мозга или ослаблением синтеза в них норадреналина [15]. Экспериментальные работы последних лет установили участие молекулярных регуляторов, синтезируемых в нервной ткани, в том числе нейронального оксида азота (NO), в реализации нейрогенных механизмов формирования АГ. Нейрональный оксид азота оказывает ингибирующее влияние на симпатическую активность как на центральном, так и на периферическом уровне. Внутрижелудочковое введение неселективного ингибитора синтеза оксида азота, блокирующего экспрессию и нейрональной (nNOS), и эндотелиальной (eNOS) NO-синтазы, активирует симпатическую нервную систему, вызывая достоверное повышение артериального давления и частоты сердечных сокращений [67]. Введение ингибиторов синтеза NO (L-NMMA, L-NAME) в область nucleus tractus solitarii, воспринимающего афферентные импульсы от баро- и хеморецепторов артерий, также вызывает повышение АД и частоты сердечных сокращений [38]. Таким образом, очевидно, что нейрональный NO непосредственно воздействует на NTS и тем самым снижает активность симпатической нервной системы.
Одним из независимых факторов риска развития эндотелиальной дисфункции в рамках АГ является возраст [16, 38]. Известно, что с возрастом активность симпатической нервной системы в ряде периферических органов, включая сердце, существенно увеличивается [68]. Таким образом, возрастассоциированная активация симпатической нервной системы может объяснять увеличение риска развития АГ при взрослении и старении.
Безусловно, патология головного мозга вследствие его ишемического, травматического, неопластического и других повреждений может изменять реализацию АД-регулирующих функций. Так, повреждение или дисфункция гипоталамуса и миндалевидных ядер может проявляться нарушением обмена серотонина и пролактина, изменением синтеза морфиноподобных нейропептидов, активацией образования эндотелина, вазопрессина и дигиталисподобного натрийуретического фактора в супраоптическом и паравентрикулярных ядрах, что вносит существенный вклад в дисрегуляцию системного АД и формирование артериальной гипертензии. Изменения состояния норадренергических нейронов (групп А1 — А2) и адренергических нейронов (группы С1), расположенных в латеральных отделах продолговатого мозга и являющихся чрезвычайно чувствительным вазопрессорным центром, также могут обусловить значительные колебания уровня АД. Установлено, что активация или торможение I1-имидазолиновых рецепторов, встроенных в мембраны этих нейронов, вызывает соответственно понижение или повышение системного АД [31].
Таким образом, детальное изучение состояния церебральной гемодинамики, внутричерепного гомеостаза и функциональной активности головного мозга на фоне нарушенной регуляции уровня системного артериального давления позволит наиболее адекватно подобрать лечение на различных стадиях артериальной гипертензии.
1. Скворцова В.И. и др. Артериальная гипертония и головной мозг // Журн. неврологии и психиатрии. — 2006. — № 10. — С. 68-76.
2. Бувальцев В.И. Дисфункция эндотелия как новая концепция профилактики и лечения сердечно-сосудистых заболеваний // ММЖ. —2001. — № 3. — С. 4-11.
3. Бурцев Е.М. Дисциркуляторная (сосудистая) энцефалопатия // Журн. невропатол. и псих. — 1998. — № 1. — С. 45-48.
4. Бурцев Е.М. Дисциркуляторная энцефалопатия // Журн. невропатол. и псих. — 2000. — № 2. — С. 33-38.
5. Верещагин Н.В., Моргунов В.А., Гулевская Т.С. Патология головного мозга при атеросклерозе и артериальной гипертензии. — М.: Медицина, 1997. — 288 с.
6. Верещагин Н.В., Калашникова Л.А., Гулевская Т.С., Миловидов Ю.К. Болезнь Бинсвангера и проблема сосудистой деменции: к столетию первого описания // Журн. неврол. и психиатр. им. С.С. Корсакова. — 1995. — 1. — 98-103.
7. Гистология / Под ред. Ю.И. Афанасьева, Н.А. Юриной. М.: Медицина, 1989. — С. 368-388; Альперн Д.Е. Патофизиологическая физиология. — М.: Медицина, 1965. — С. 146-160.
8. Гусев В.И., Скворцова В.И. Ишемия головного мозга. — М.: Медицина, 2001. — 328 с.
9. Карлов В.А. и др. Дисциркуляторная энцефалопатия у больных артериальной гипертензией // Журн. невропатол. и псих. — 1997. — № 5. — С. 15-17.
10. Шпрах В.В. и др. Дисциркуляторная энцефалопатия у больных сахарным диабетом / // Неврологич. журнал. — 1998. — № 6. — С. 32-34.
11. Евстигнеев В.В., Юршевич Е.А., Бузуева О.А. Дисциркуляторная энцефалопатия // Медицина. — 2001. — № 1. — С. 26-29.
12. Ганнушкина И.В., Лебедева Н.В. Гипертоническая энцефалопатия. — М.: Медицина, 1987. — 223 c.
13. Карпов Ю.А. Европейские рекомендации по артериальной гипертензии — главное событие 2007 года // РМЭИ. — 2007. — № 12. — С. 1405-1408.
14. Горбачев В.В. и др. Клиническая кардиология: Руководство для врачей. — Мн.: Книжный дом, 2007. — 864 с.
15. Кушаковский М.С. Гипертоническая болезнь (эссенциальная гипертония): причины, механизмы, клиника, лечение. — СПб.: СОТИС, 1995. — 320 с.
16. Лелюк В.Г., Лелюк С.Э. Ультразвуковая ангиология. — М.: Реальное время, 2003. — С. 37.
17. Медведев А. В. Патогенез сосудистой деменции // Журн. невропатол. и псих. — 1995. — № 5. — С. 95-100.
18. Молоков Д.Д., Бурцев Е.М. Констрикторные реакции мозговых сосудов в патогенезе дисциркуляторной энцефалопатии // Журн. невропатол. и псих. — 1996. — № 5. — С. 64-67.
19. Морман Д., Хеллер Л. Физиология сердечно-сосудистой системы. — СПб.: Питер, 2000. — С. 102-218.
20. Верещагин Н.В. и др. Оценка цереброваскулярного резерва при атеросклеротическом поражении сонных артерий // Журн. невропатол. и псих. — 1999. — № 2. — С. 57-64.
21. Постнов Ю.В. К истокам первичной гипертензии: подход с позиций биоэнергетики // Кардиология. — 1998. — № 12. — С. 11-48.
22. Чазов Е.И. и др. Рациональная фармакотерапия сердечно-сосудистых заболеваний: руководство для врачей. — М.: Литтерра, 2005. — 972 с.
23. Шабалин В.Н. и др. Руководство по геронтологии. — М.: Цитадель-трейд, 2005. — 800 с.
24. Сіренко Ю. Артеріальна гіпертензія: діагностика, лікування та профілактика у різних категорій пацієнтів // Ліки України. — 2004.
25. Скворцова В.И., Соколов К.В.,. Шамалов Н.А. Артериальная гипертония и цереброваскулярные нарушения // Журнал неврологии и психиатрии им. С.С. Корсакова.
26. Скворцова В.И., Соколов К.В., Шамалов Н.А. Артериальная гипертония и цереброваскулярные нарушения // Журн. неврологии и психиатрии. — 2006. — № 11. — С. 57-65.
27. Скворцова В.И. Инсульт. Участие апоптоза в формировании инфаркта мозга // Журн. неврол и психиатр им. С.С. Корсакова (приложение). — 2001. — 2. — 12-9.
28. Физиология человека / Под ред. В.М. Покровского, Г.Ф. Коротько. — М.: Медицина, 1998. — С. 363-396.
29. Хвеледиани Д.Я. Ультраструктурные изменения артериол при гипертонии у человека // Кардиология. — 1977. — 17 (2). — 100-5.
30. Чазова И.Е. Инсульт. Лечение артериальной гипертонии как профилактика ишемического инсульта // Журн. неврол. и психиатр. им. С.С. Корсакова (приложение). — 2001. — 3. — 3-7.
31. Шевченко О.П. и др. Артериальная гипертония и церебральный инсульт. — М.: Реафарм, 2001. — 187 с.
32. Шмырев В.И., Гулевская Т.С., Попова С.А. Гипертоническая дисциркуляторная энцефалопатия. Нейровизуализация и патоморфология. — М.: Глав. научно-исследовательский вычислительный центр Управления делами президента РФ, 2001. — 136 с.
33. Arvin В., Neville L.F., Barone F.C., Feuerstein G.Z. // Neurosci Biobehav Rev. — 1996. — 20, 3. — 445-452.
34. Baezner H., Daffertshofer M. Subcortical vascular encephalopathy // Ther. Umschr. — 2003. — 60, 9. — 541-552.
35. Bayliss W.M. On the local reactions of the arterial wall to changes in internal pressure // J. Physiol. — 1902. — V. 28. — P. 200-231.
36. Collins R. et al. Blood pressure, stroke, and coronary heart disease, part 2: short-term reductions in blood pressure: overview of randomised drug trials in their epidemiological context // Lancet. — 1990. — Vol. 335. — P. 827-838.
37. Chillon J.M., Baumbach G.L. Effects of an angiotensin-converting enzyme inhibitor and a beta-blocker on cerebral arteriolar dilatation in hypertensive rats // Hypertension. — 2001. — 37. — 1388-93.
38. Chowdhary S., Townend J.N. Nitric oxide and hypertension: not just an endothelium derived relaxing factor! // J. Hum. Hypertens. — 2001. — 15, 4. — 219-227.
39. Clark R.S., Kochanek P.M., Schwan M.A. et al. Inducible nitric oxide synthase expression in cerebrovascular smooth muscle and neutrophils after traumatic brain injury in immature rats // Pediat. Res. — 1996. — 39, 5. — 784-790.
40. Puddu G.M. et al. Clinical aspects and pathogenetic mechanisms of cognitive impairment in arterial hypertension // Minerva. Cardioangiol. — 1996. — Vol. 44, № 6. — P. 285-297.
41. Cupini L.M., Pasqualetti P., Diomedi M. et al. Carotid artery intima-media thickness and lacunar versus nonlacunar infarcts // Stroke. — 2002. — 33. — 689.
42. De Vries H.E., Kuiper J., De Boer A.G. et al. The blood-brain barrier in neuroinflammatory diseases // Pharmacol. Rev. — 1997. — 49, 2. — 143-155.
43. De Vries H.E., Moor A.C., Blom-Roosemalen M.C. et al. Lymphocyte adhesion to brain capillary endothelial cells in vitro // J. Neuroimmunol. — 1994. — 52 (1). — 1-8.
44. Castillo V. et al. Etiology and mechanism in cerebral infarction // Schweiz. Med. Wochenschr. — 1996. — Vol. 126, № 12. — P. 489-492.
45. Ferrario C.M. Use of angiotensin II receptor blockers in animal models of atherosclerosis // Am. J. Hypertens. — 2002. — 15. — 9-13.
46. Folkow B. Description of the Myogenic Hypothesis // Circ. Res. — 1964. — V. 14 (suppl. 1). — P. 1279-1287.
47. Fujishima M., Tsuchihashi T. Hypertension and dementia // Clin. Exp. Hypertens. — 1999. — Vol. 21, № 5–6. — P. 927-935.
48. Gibbons G.H., Dzau V.J. The emerging concept of vascular remodeling // New Engl. J. Med. — 1994. — № 330. — Р. 1431-1438.
49. Giulian D. Reactive glia as rivals in regulating neuronal survival // Glia. — 1993. — 7, 1. — 102-110.
50. Gorelick P.B. New horizons for stroke prevention: PROGRESS and HOPE // Lancet Neurol. — 2002. — 1. — 149-56.
51. Hamson D.G. Endothelial function and oxidant stress // Clin. Cardiology. — 1997. — № 20 (SII). — Р. 11-17.
52. Hawkins C.P., Munro P.M., MacKenzie F. et al. Duration and selectivity of blood-brain barrier breakdown in chronic relapsing experimental allergic encephalomyelitis studied by gadolinium-DTPA and protein markers // Brain. — 1990. — 113. — 365-378.
53. Hock F.J., 1995; Karpati S. et al., 2002.
54. Intengan H.D., Schiffrin E.L. Structure and mechanical properties of resistance arteries in hypertension: role of adhesion molecules and extracellular matrix determinants // Hypertension. — 2000. — 36. — 312-8.
55. Provinciali L. et al. Investigation of cerebrovascular reactivity using transcranial Doppler sonography. Evaluation and comparison of different methods // Funct. Neurol. — 1990. — Vol. 5, № 1. — P. 33-41.
56. Kontos H.A. Oxygen radicals in cerebral vascular injury // Circul. Res. — 1985. — 57 (4). — 508-16.
57. Mac Mahon S., Peto R., Cutler J. et al. Blood pressure, stroke and coronary heart disease. Part 1. Prolonged differences in blood pressure: prospective observational studies corrected for regression dilution bias // Lancet. — 1990. — 335. — 765-74.
58. MacMahon S. Blood pressure and the risk of cardiovascular disease // N. Engl. J. Med. — 2000. — 342. — 50-2.
59. McGeer P.L., Kawamala Т., Walker D.G. et al. // Glia. — 1993. — 7. — 84-92.
60. Nag S., Robertson D.M., Dinsdale H.B. Cerebral cortical changes in acute experimental hypertension: An ultrastructural study // Lab. Invest. — 1977. — 36 (2). — 150-61.
61. Nag S. Cerebral changes in chronic hypertension: combined permeability and immunohistochemical studies // Acta Neuropathol. (Berl.). — 1984. — 62 (3). — 178-84.
62. Nag S. Cerebral endothelial plasma membrane alterations in acute hypertension // Acta Neuropathol. (Berl.). — 1986. — 70 (1). — 38-43.
63. Nag S. Localisation of calcium-activated adenosine-triphosphatase (Ca2+-ATPase) in intracerebral arterioles in acute hypertension // Acta Neuropathol. (Berl.). — 1988. — 75 (6). — 547-53.
64. Nagai Y., Kitagawa K., Matsumoto M. Implication of earlier carotid atherosclerosis for stroke and its subtypes // Prev. Cardiol. — 2003. — 6 (2). — 99-103.
65. Nawroth P.P., Stem D.M. Modulation of endothelial cell hemostatic properties by tumor necrosis factor // Exp. Med. J. — 1986. — 163. — 740-5.
66. Oggunniyi A., Talabi O. Cerebrovascular complications of hypertension // Niger. J. Med. — 2001. — 10, 4. — 158-161.
67. Sakuma I., Togashi H., Yoshioka M. et al. NG-methyl-L-arginine, an inhibitor of L-arginine-derived nitric oxide synthesis, stimulates renal sympathetic nerve activity in vivo. A role for nitric oxide in the central regulation of sympathetic tone? // Circ. Res. — 1992. — 70. — 607-611.
68. Seals D.R., Dinneno F.A. — 2004. — 287. — 5. — 1895-1905.
69. Siren A.-L., McCarron R.M., Liu Y. et al. In: Microcirculatory stasis in the brain / Ed. by M. Tomita, G. Mchedlishvili, W.I. Rosenblum, W.-D. Heiss, Y. Fukuuchi. — Amsterdam: Excerpta Medica, 1993. — 169-175.
70. Strandgaard S., Paulson O.B. Cerebrovascular damage in hypertension // J. Cardiovasc. Risk. — 1995. — Vol. 2, № 1. — P. 9-34.
71. Strandgaard S. Hypertension and stroke // J. Hypertens. — 1996. — Vol. 14, № 3. — P. 7-23.
72. Bogousslavsky J. et al. Stroke subtypes and hypertension. Primary hemorrhage vs infarction, large- vs small-artery disease // Arch. Neurol. — 1996. — Vol. 53, № 3. — P. 265-269.
73. Tomita M., Fukuuchi Y., T erakawa S. Differential behavior of glial and neuronal cells exposed to hypotonic solution // Acta Neurochir. — 1994. — 1 (60). — 31-3.
74. Vemmos K.N., Tsivgoulis G., Spengos K. et al. Common carotid artery intima-media thickness in patients with brain infarction and intracerebral haemorrhage // Cerebrovasc. Dis. — 2004. — 17 (4). — 280-6.
75. Rodgers A., Mac Mahon S., Gamble G. et al. Blood pressure and risk of stroke in patients with cerebrovascular disease. The United Kingdom Transient Ischaemic Attack Collaborative Group // BMJ. — 1996. — 313. — 147.