
Международный неврологический журнал 4 (42) 2011
Вернуться к номеру
Необычная комбинация ювенильной миастении и миодистрофии. Катамнестическое наблюдение
Авторы: Кушнир Г.М., Самохвалова В.В., Ларина Н.В., Крымский государственный медицинский университет им. С.И. Георгиевского, г. Симферополь, Украина
Рубрики: Неврология
Версия для печати
В статье представлено катамнестическое наблюдение пациента с ювенильной миастенией. Обсуждается присоединение симптомов миодистрофии («миопатизация») при длительном течении ювенильной миастении. Высказывается предположение о своеобразной комбинации ювенильной миастении и оригинального варианта миодистрофии.
Ювенильная миастения, миодистрофия.

Миастения — аутоиммунное нервно-мышечное заболевание, клинически характеризующееся слабостью и патологической утомляемостью поперечнополосатой мускулатуры и по общепринятым представлениям связанное с повреждением ацетилхолиновых рецепторов постсинаптической мембраны поперечнополосатых мышц специфическими комплементфиксирующими антителами [1–3, 5, 6].
Ювенильной принято называть миастению, дебютирующую в возрасте до 18 лет. У детей первые симптомы миастении никогда не возникают до шестимесячного возраста, у 75 % из них симптоматика появляется после 10 лет. Чаще дебют заболевания наблюдается у мальчиков в препубертатном возрасте, при этом отмечаются лишь глазные симптомы и отсутствуют антитела к ацетилхолиновым рецепторам. При локальной форме миастении наряду с глазодвигательной мускулатурой может вовлекаться лицевая и бульбарная. Однако расстройства речи и глотания, дыхательные нарушения отсутствуют. Глазная форма миастении имеет прогрессирующее или ремиттирующее течение. При ремиттирующем течении обострения варьируют по тяжести и продолжаются от нескольких недель до нескольких лет. Длительные ремиссии отмечаются у 20 % больных. Спонтанные ремиссии более характерны при дебюте миастении в препубертатный период, нежели в постпубертатный [6].
У детей с генерализованной формой заболевания распространенная мышечная слабость и патологическая утомляемость может обнаруживаться через 1 год после появления глазных симптомов. Зачастую наблюдаются нарушения речи и глотания, трудности при жевании и утомляемость в конечностях. Для генерализованной формы миастении спонтанные ремиссии не характерны. Дети, имеющие данную форму миастении, имеют высокий риск развития аутоиммунных заболеваний, особенно тиреоидита и коллагенозов. Тимома у данного контингента пациентов наблюдается менее чем в 5 % случаев. Примерно у 10 % больных постепенно формируется мышечная атрофия. При исследовании неврологического статуса сухожильные рефлексы обычно остаются сохранными, однако при значительном ослаблении соответствующей мышцы они снижаются [5, 6].
Приводим описание клинического случая.
Больной К., 38 лет, инвалид второй группы, обратился с жалобами на осиплость голоса, поперхивание при еде, слабость лицевых, жевательных мышц, слабость кистей.
Из анамнеза известно, что заболел в 10 лет, когда появилась общая мышечная слабость, больше в ногах («не держали ноги»), приводящая к падениям. Упав, ребенок не мог самостоятельно подняться, однако после кратковременного отдыха в течение 10–15 минут подняться все же удавалось. Мышечная слабость меньше беспокоила в первую половину дня и нарастала к вечеру.
В 11-летнем возрасте ребенок консультировался в г. Москве в Миастеническом центре профессором Б.М. Гехтом, где на основании данных анамнеза, клинической картины, положительной прозериновой пробы поставлен диагноз миастении, генерализованной формы. По рекомендации принимал оксазил, верошпирон с положительным эффектом.
В 12-летнем возрасте появилось опущение век, более выраженное к концу дня. В 15 лет больному была проведена тимэктомия. После операции слабость в мышцах конечностей не беспокоила, но постепенно присоединились осиплость голоса и поперхивание при еде. Продолжал принимать оксазил по 1 таблетке 3 раза в день без эффекта, вследствие чего вскоре отказался от приема препарата.
Постепенно стал замечать нарастающую слабость мимической мускулатуры, жевательных мышц, продолжали беспокоить осиплость голоса и поперхивание при еде. Примерно в 25-летнем возрасте наблюдалась стабилизация симптоматики. В это же время обратил внимание на слабость в кистях, затрудняющую их разгибание. У невропатолога по настоящее время не наблюдался.
Наследственность не отягощена. Соматическими заболеваниями не страдал. Был женат, имеет здоровую дочь.
Объективно: общее состояние больного удовлетворительное. В соматическом статусе пациента патологии не выявлено.
Неврологический статус: сознание ясное, ориентирован, адекватен. Общемозговой и менингеальной симптоматики нет. Зрачки одинаковой величины, фотореакция и корнеальные рефлексы живые. Асимметричный птоз, более выражен слева, диплопия при взгляде влево и вверх. Отмечается нарастание птоза при нагрузочных пробах. Движения глазных яблок не ограничены. Слабость и атрофия мимических, жевательных мышц (рис. 1). Атрофия мышц языка, затруднено высовывание языка. Глоточный рефлекс отсутствует. Выражены дисфония, дисфагия, дизартрия.
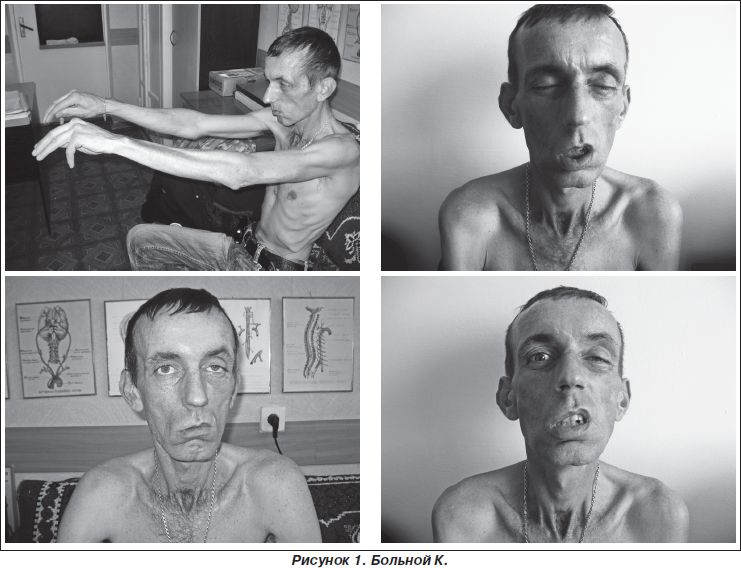
Обращает на себя внимание генерализованная мышечная гипотрофия, более выраженная в мышцах плечевого пояса, руках, туловище (рис. 1). Мышечная сила в проксимальных отделах верхних конечностей, в нижних конечностях сохранена. Выявлены парезы сгибателей и разгибателей кистей и пальцев, там же более выраженные атрофии мышц, отсутствие карпорадиальных и снижение сухожильных рефлексов с m.biceps и m.triceps, сгибательные контрактуры кистей. Сухожильные рефлексы на нижних конечностях живые. Патологических рефлексов нет. Чувствительных нарушений не выявлено.
Дополнительные обследования: общеклинические и биохимические анализы крови и мочи, электролиты, сахар крови без патологии. Функция щитовидной железы не нарушена. Креатинфосфокиназа 706 ед/л (норма 38,0–174,0). Лактат 1,92 (норма 0,5–2,2).
При проведении прозериновой пробы наблюдалось уменьшение птоза век. Другой положительной динамики выявлено не было.
На глазном дне: ДЗН деколорированы темпорально, границы четкие, расширенная экскавация, артерии сужены; макулярная область и видимая периферия без особенностей.
Заключение: простой астигматизм ОD. Смешанный астигматизм OS. Ангиопатия сетчатки обоих глаз.
ЭКГ: ритм синусовый, электрическая позиция сердца не отклонена.
УЗИ сердца: пролапс митрального клапана до 6,5 мм, дополнительная хорда в полости левого желудочка. Сократительная способность миокарда сохранена.
ЭМГ-исследование состояния нервно-мышечной передачи при ритмической стимуляции срединных нервов с частотой 3 Гц (декремент-тест) выявило резкое снижение амплитуды М-ответов, составляющее 40 % (норма — до 5 %). При стимуляционной ЭМГ срединных нервов выявлено снижение амплитуды М-ответа до 1,76 мВ (норма — 3,5–8,0 мВ). Скорость проведения нервного импульса — 60 м/с (норма — 50–70 м/с).
Обсуждение
У пациента наряду с клинической картиной ювенильной миастении (начало заболевания в детском возрасте с генерализованной мышечной слабости и патологической утомляемости и положительной реакции на антихолин-эстеразные препараты, с присоединением в последующем слабости лицевой, бульбарной, скелетной мускулатуры; относительная стабильность симптоматики на протяжении длительного времени; данные ЭМГ, частично положительные результаты прозериновой пробы) имеется симптоматика, характерная для миодистрофии (медленно нарастающая слабость и атрофии мышц лица, глотки, языка, повышение уровня КФК в сыворотке крови) [3, 6]. Однако у больного отсутствует поражение проксимальных мышц конечностей при наличии парезов дистальных отделов верхних конечностей, осложнившихся контрактурами.
Течение заболевания также представляет интерес. Начальная генерализованная слабость в детском возрасте, хорошо откликающаяся на антихолинэстеразные препараты, после оперативного лечения исчезает и постепенно появляется нарастающая слабость лицевой, бульбарной мускулатуры с атрофиями дистальных отделов верхних конечностей.
Мышечные атрофии могут появляться при длительно протекающей миастении, однако они не бывают столь выраженными.
Дифференциальная диагностика проводилась с митохондриальной энцефаломиопатией (синдромом Кернса — Сейра), лице-плече-лопаточной формой миодистрофии Ландузи — Дежерина.
У нашего больного, кроме прогрессирующей наружной офтальмоплегии, отсутствовали другие симптомы, характерные для митохондриальной энцефаломиопатии Кернса — Сейра (пигментная ретинопатия, кардиомиопатия с нарушением проводящей системы и развитием полной атриовентрикулярной блокады). Диагнозу миодистрофии Ландузи — Дежерина противоречит дистальная локализация парезов в конечностях. Предположение о комбинации миопатии с полимиозитом (комплекс миастения — полимиозит) может быть отвергнуто на основании длительного течения заболевания с отсутствием экстраневральной симптоматики.
В доступной нам литературе не встречались катамнестические наблюдения за больными с ювенильной миастенией. В нашем случае речь идет либо о типичном катамнезе ювенильной миастении, которая при длительном течении приобретает черты миодистрофии («миопатизируется»), либо о своеобразной комбинации ювенильной миастении и оригинального варианта миодистрофии.
В практической деятельности врач-невролог периодически встречается с пациентами, ведущим синдромом заболеваний которых являются глазодвигательные и бульбарные нарушения. Данный симптомокомплекс характерен для достаточно большого спектра заболеваний. Глубокое знание этих нозологических форм, критериев диагностики позволяет максимально приблизиться к постановке правильного диагноза и определению тактики ведения пациента.
1. Гехт Б.М. Синдромы патологической мышечной утомляемости. — М.: Медицина, 1974. — 200 c.
2. Гехт Б.М., Санадзе Л.Г. Миастения: диагностика и лечение // Неврологический журнал. — 2003. — № 1. — С. 8-11.
3. Евтушенко С.К., Шаймурзин М.Р., Евтушенко О.С. и др. Ранняя клинико-инструментальная диагностика и терапия быстро- и медленнопрогрессирующих мышечных дистрофий и амиотрофий // Международный неврологический журнал. — 2007. — № 4 (14). — С. 14-30.
4. Максимова Н.Р., Коротов М.Н., Николаева И.А. и др. Клинические и молекулярно-генетические аспекты окулофарингеальной миодистрофии в Республике Саха (Якутия) // Генетика и патология / Под ред. В.П. Пузырева. — Томск: Печатная мануфактура, 2007. — Вып. 8. — С. 160-161.
5. Хосе Биллер. Практическая неврология. Лечение. — М.: Мед. лит. — 2008. — 416 с.
6. Яхно Н.Н., Штульман Д.Р. Болезни нервной системы: В 2 т. — М.: Медицина, 2003. — Т. 1. — 743 с.