
Международный неврологический журнал 6 (44) 2011
Вернуться к номеру
Болезнь Паркинсона: патогенетические аспекты лекарственной терапии и клинического течения
Авторы: Карабань И.Н., ГУ «Институт геронтологии им. Д.Ф. Чеботарева НАМН Украины», г. Киев
Рубрики: Неврология
Версия для печати
Приведены данные о классификации паркинсонизма, обсуждены современные представления о патогенезе болезни Паркинсона, стадийности течения заболевания, принципах терапии. К основным классам препаратов патогенетического и симптоматического действия относят агонисты дофаминовых рецепторов, ингибиторы МАО-В, блокаторы глутаматных рецепторов, леводопа-содержащие препараты, ингибиторы КОМТ (COMT inhibitors), холиноблокаторы. Обсуждаются новые технологии лечения: duodopa-помпа, DBS, апоморфин-помпа.
Паркинсона, патогенетическое лечение, основные классы противопаркинсонических препаратов.
Болезнь Паркинсона (БП) — хроническое прогрессирующее заболевание центральной нервной системы (ЦНС), относящееся к классу дофаминзависимых нейродегенераций [1, 2, 8, 28].
Существуют три основные этиопатогенетические формы паркинсонизма:
— первичный (идиопатический) паркинсонизм, который включает в себя непосредственно болезнь Паркинсона и ювенильный паркинсонизм;
— вторичный (симптоматический) паркинсонизм, который в зависимости от этиологического фактора может быть постэнцефалическим, лекарственным, сосудистым, токсическим, травматическим;
— паркинсонический синдром при различных формах мультисистемной дегенерации (болезнь Вильсона — Коновалова, хорея Гентингтона, кортико-базальная дегенерация и др.) (табл. 1).
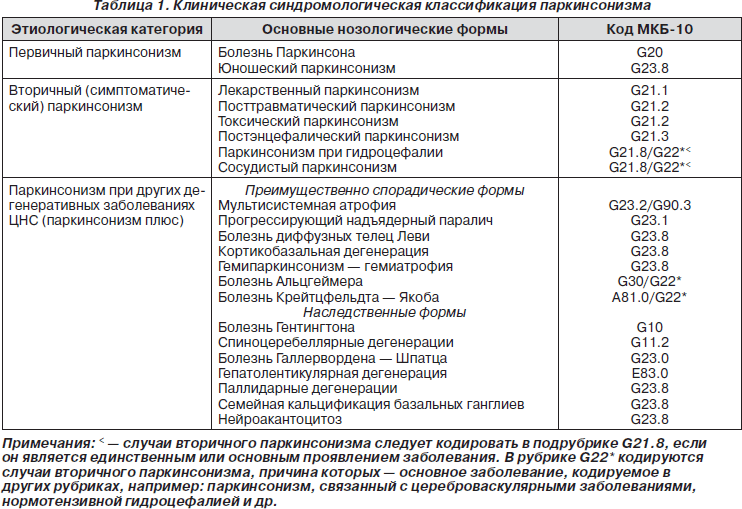
Болезнь Паркинсона (или идиопатический паркинсонизм) выделена в отдельную нозологическую форму в отличие от симптоматического паркинсонизма (вторичный паркинсонизм) и заболеваний, в клинической картине которых имеются отдельные паркинсонические симптомы, — мультисистемная дегенерация с синдромом «паркинсонизм плюс» (табл. 1). Имея общий синдром двигательных нарушений, эти заболевания значительно отличаются по этиологии и патогенезу и не чувствительны к классической противопаркинсонической терапии. Единственным надежным критерием, подтверждающим клинический диагноз БП, считается специфическая реакция на леводопу — положительный леводопа-тест, результатом которого является ответный регресс двигательных нарушений.
Типичным возрастом манифестации клинических симптомов БП считают 45–52 года. Важно отметить, что к этому времени происходит значительное снижение функциональной активности дофаминсинтезирующих нейронов нигростриатной системы, что позволяет рассматривать БП и как ассоциированную с возрастом патологию [5, 7, 17]. Фактором риска развития БП является возрастная экстрапирамидная недостаточность (ЭПН), симптомы которой четко проявляются по мере старения человека [1, 2]. У 7,5–10 % лиц старше 60 лет возрастная ЭПН трансформируется в БП [5]. По данным мировой статистики, частота БП составляет от 60 до 187 [20], а в Украине — 133 на 100 тыс. населения (С.П. Московко, 2005).
Патогенез
По современным данным, БП считают болезнью нейромедиаторного обмена. Специфической биохимической особенностью заболевания является недостаточность продукции дофамина (ДА) в базальных ганглиях и развитие ДОФА-дефицитарного нейромедиаторного дисбаланса. Клинические признаки БП появляются при потере не менее 70 % ДА в стриатуме (хвостатом ядре и скорлупе). Причиной возникающих двигательных нарушений является уменьшение тормозного дофаминового контроля, осуществляемого нигростриатной системой, и гиперактивация стриатных холинергических нейронов [1, 14]. Недостаточность ДА-трансмиссии, развивающаяся вследствие дегенерации значительной части дофаминсинтезирующих нейронов, приводит к повышению активности ферментов катаболизма ДА-моноаминоксидазы Б (МАО-Б) и катехол-О-метилтрансферазы (КОМТ), к изменению функциональных взаимоотношений между ДА и возбуждающим медиатором глутаматом. В свою очередь, гиперактивация глутаматных рецепторов усиливает кальциевый ток и накопление кальция в ДА-нейроне, способствуя индукции механизмов повреждения и гибели нигростриатных нейронов.
Один из ведущих факторов, усугубляющих некроз и апоптоз ДА-нейронов, — так называемый окислительный стресс [2, 25]. Установлено, что при БП усиливается свободнорадикальное окисление. Цитотоксичность свободных радикалов связана с ингибированием сульфгидрильных ферментов, полимеризацией протеинов, активацией процесса перекисного окисления липидов, токсически действующего на клеточные митохондрии, белки, нуклеиновые кислоты и нейрональные мембраны. Высокий риск воздействия гидроксильных радикалов на ДА-нейроны связан с образованием гидроксипероксида при реакции окисления ДА с помощью МАО-Б [23].
Важное клиническое значение приобрели открытия, связанные с МРТР-нейротоксином, избирательно повреждающим ДА-нейроны черной субстанции [25]. Было показано, что ингибиторы МАО-Б защищают экспериментальных животных от БП, вызванной МРТР, а следовательно, могут тормозить прогрессирование болезни у человека [18, 24].
Морфологические изменения при БП локализуются не только в нигростиатуме, но и в других отделах головного мозга (бледном шаре, ядрах среднего мозга, таламусе, голубом пятне). Считают, что двигательные нарушения у таких больных обусловлены не только функциональными изменениями нейронных ансамблей в этих регионах мозга, но и нарушением интегративных взаимоотношений между соответствующими структурами ствола, подкорковых образований и коры.
Важно отметить, что синтез ДА осуществляется не в аксоне, а в теле ДА-нейрона, расположенного в черной субстанции, где происходят поэтапные ферментативные превращения в цепи «фенилаланин тирозин L-ДОФА дофамин» [1, 2, 19]. Здесь ДА «складируется» в гранулы хранения и по мере необходимости транспортируется к микровезикулам, разветвления которых представляют собой пресинаптическую часть ДА-синапса. Высвобождение ДА в синаптическую щель происходит в виде кванта медиатора под влиянием нервного импульса. Взаимодействуя с ДА-рецептором постсинаптической мембраны, ДА вызывает ее деполяризацию, после чего 80 % выделившегося медиатора удаляется в пресинаптическое пространство по механизму обратного захвата (re-uptake), а оставшаяся часть ДА инактивируется с помощью МАО-Б и КОМТ.
Знание этих деталей синаптической передачи необходимо для медикаментозного управления отдельными этапами метаболизма нейромедиаторов при БП.
Болезнь Паркинсона традиционно рассматривается как заболевание, преимущественно поражающее моторную сферу. Классические двигательные проявления болезни Паркинсона — гипокинезия, ригидность, тремор покоя — вызваны дегенерацией дофаминергических нейронов компактной части черной субстанции и возникающим в силу этого дефицитом дофамина в стриатуме. Но, помимо моторных симптомов, в клинической картине болезни Паркинсона присутствуют и немоторные проявления. Более того, по мере прогрессирования заболевания некоторые из них приобретают доминирующее клиническое значение, оказывая негативное влияние на качество жизни пациентов, приводя к их инвалидизации и сокращая продолжительность жизни. Немоторные проявления болезни Паркинсона включают вегетативные, психические, диссомнические, сенсорные и некоторые другие нарушения [4, 8, 9].
Большинство немоторных проявлений появляются и нарастают по мере прогрессирования заболевания — параллельно с усугублением двигательных расстройств. Но некоторые немоторные проявления, такие как нарушения обоняния, запоры, депрессия, расстройство поведения во сне с быстрыми движениями глаз (БДГ), болевые синдромы, возникают до развития классических моторных симптомов болезни Паркинсона. В связи с этим говорят о премоторной стадии болезни Паркинсона. Отсутствие специфических симптомов делает клиническую диагностику болезни Паркинсона на премоторной стадии практически невозможной. Тем не менее обследование пациентов с подобными нарушениями с помощью функциональных методов нейровизуализации и некоторых других инструментальных методов, особенно если у них есть родственники, страдающие болезнью Паркинсона, — перспективный путь к максимально раннему выявлению болезни Паркинсона.
Достигнутые в последние десятилетия успехи в увеличении продолжительности жизни больных БП привели к тому, что немоторные проявления все чаще отмечаются на поздних стадиях болезни, особенно у пожилых лиц. По данным Сиднейского мультицентрового исследования, включавшего наблюдение за больными БП в течение 20 лет, немоторные симптомы, обычно резистентные к леводопе, инвалидизировали пациентов в большей степени, чем основные двигательные проявления заболевания [2, 10].
Новое понимание механизмов развития и роли немоторных проявлений в структуре болезни Паркинсона несет концепция H. Braak и соавт. (2003), согласно которой дегенеративный процесс не ограничивается компактной частью черной субстанции, а последовательно вовлекает большое количество мозговых структур. H. Braak и соавт. выделили 6 стадий развития патологического процесса при болезни Паркинсона [10]. Первая стадия характеризуется дегенерацией обонятельной луковицы и переднего обонятельного ядра, которая клинически может проявиться нарушением обоняния. Вторая стадия характеризуется вовлечением ядер ствола мозга, контролирующих аффективные, вегетативные функции, цикл сна и бодрствования, и может проявляться расстройством поведения во сне с БДГ, депрессией, запорами. Классические моторные проявления болезни Паркинсона появляются только на 3-й и 4-й стадиях, что связано с распространением дегенеративного процесса на черную субстанцию. В финальных 5-й и 6-й стадиях тельца Леви появляются в лимбических структурах и коре головного мозга, что приводит к развитию когнитивных, поведенческих и психотических расстройств [10, 11].
Известно, что значительная часть немоторных проявлений резистентна к препаратам леводопы, что указывает на их связь с дисфункцией недофаминергических систем: норадренергических, серотонинергических, холинергических и др. Тем не менее нередко немоторные проявления возникают или усиливаются в связи с действием противопаркинсонических средств.
При оценке темпа развития болезни Паркинсона как хронического прогрессирующего заболевания принято определять стадии болезни. Существуют различные классификации стадийности течения БП. Н.Б. Маньковский и соавт. (1982) выделяют три степени заболевания, подразделяя каждую из них на подгруппы А и Б в соответствии с клинико-функциональными проявлениями прогредиентности течения БП. Начальным проявлениям заболевания соответствует I степень. Степень IА характеризуется нерезко выраженным амиостатическим симптомокомплексом, иногда небольшим дрожанием (или только дрожанием). Нарушения двигательных функций не наблюдаются. Эта степень соответствует экстрапирамидной недостаточности. Для степени IБ характерны замедленность активных движений, более выраженное дрожание, начальные вегетативные нарушения. При степени II имеются умеренно выраженные изменения. Степень IIА характеризуется повышением мышечного тонуса в двух или более конечностях, заметным замедлением активных движений или почти постоянным дрожанием, выраженной вегетативной симптоматикой; могут нарушаться когнитивные функции. Такие больные еще в состоянии выполнять свою профессиональную работу, если она не связана с тонкими и точными движениями. При степени IIБ симптоматика еще более нарастает, часто нарушена походка. Больные уже не могут работать, однако полностью себя обслуживают в быту, выполняют домашнюю работу. Тяжелой форме заболевания соответствует III степень. Для степени IIIА характерны выраженная мышечная ригидность или дрожание, брадикинезия, нарушение статики и походки, резкая вегетативная дисфункция, значительные изменения высшей нервной деятельности, соматическая патология. Возможность самообслуживания ограничена. При степени IIIБ наблюдаются почти полная обездвиженность, акинезия; больные прикованы к постели, самообслуживание, даже элементарное, невозможно, необходим постоянный посторонний уход.
В настоящее время используют международную шкалу оценки стадийности БП M. Hoehn и M. Yahr (1967) в модификации Lindvall (1987), которая по своей градации полностью совпадает с функциональной классификацией Н.Б. Маньковского и соавт. (1982) (табл. 2).

Важное значение для объективизации этапов течения болезни Паркинсона приобретает показатель скорости прогрессирования заболевания. Н.В. Федорова (2002) предлагает выделять быстрый темп, который характеризуется сменой стадий в течение 2 лет, умеренный — от 3 до 5 лет и медленный — более 5 лет.
В зависимости от преобладания ведущего симптома в триаде двигательных нарушений определяют клиническую форму болезни Паркинсона: акинетико-ригидно-дрожательную, ригидно-акинетико-дрожательную или дрожательно-акинетико-ригидную.
Критерии включения/исключения. Существует более тридцати неврологических синдромов, сходных по клинической картине с болезнью Паркинсона. Поэтому для того чтобы правильно поставить диагноз заболевания и назначить эффективное патогенетически значимое лечение, требуется консультация врача, имеющего профильную специализацию по экстрапирамидной патологии. Вначале ставят синдромальный диагноз паркинсонизма, при этом учитывают международные критерии включения/исключения (A. Hughes, 1992; UK Brain Bank Criteria), а также возраст больного и сопутствующую патологию [15].
На сегодняшний день существуют современные методы диагностики болезни Паркинсона, такие как клинический леводопа-тест, позитронно-эмиссионная томография (ПЭТ), электромиография (ЭМГ).
Для постановки леводопа-теста в течение пяти дней больной должен принимать леводопасодержащие препараты (синемет, наком или мадопар) в суточной дозе 200–250 мг, разделенной на три приема в течение дня, после чего регистрируют степень изменения (уменьшения) показателей двигательных функций с помощью специализированной шкалы Unified Parkinson’s Disease Rating Scale (UPDRS; III часть, моторная активность) [13]. Если ожидаемый ответ на леводопа-тестирование не наблюдается, то с наибольшей вероятностью заболевание нельзя расценивать как болезнь Паркинсона. Одним из достоверных методов диагностики заболевания считается ПЭТ, которая позволяет выявлять снижение активности дофаминергической системы в ответ на внутривенное введение Fluorodopa еще на доклинической стадии. Метод, который доступен для применения в неврологической клинике, — ЭМГ. Важным преимуществом этого метода является его информативность для выявления субклинических и начальных проявлений заболевания, а также при оценке эффективности противопаркинсонических препаратов.
Принципы терапии
Традиционно лекарственные препараты для лечения болезни Паркинсона рассматривали как исключительно симптоматическую терапию. Ранее существовало мнение, что лечение на начальных стадиях БП не следует начинать до тех пор, пока симптомы паркинсонизма не станут влиять на социальную, бытовую и профессиональную активность больного [8, 12]. Однако достижения в изучении патогенеза БП и появившиеся новые эффективные препараты внесли соответствующие изменения в стратегию и тактику патогенетической терапии заболевания. Недавние исследования с применением нейровизуализационных методов показали, что латентный период от начала гибели нигростриатных нейронов до появления первых двигательных симптомов БП составляет от 6 до 7 лет [12]. Нарастание двигательных нарушений на ранней стадии БП происходит относительно быстро: за 1-й год наблюдения оценка по шкале UPDRS увеличивается на 8–10 баллов, что сопровождается значительным ухудшением качества жизни [11].
Некоторые авторы считают, что раннее назначение симптоматической терапии оказывает благоприятное влияние в отношении ближайшего и долгосрочного улучшения двигательных симптомов, качества жизни и уменьшения вероятности раннего появления моторных флуктуаций и дискинезий [1, 8, 26]. В настоящее время появились теоретические и практические основания для пересмотра традиционного взгляда на сроки начала дофаминергической терапии при БП. Начало лечения сразу после диагностики заболевания рассматривается как более перспективная и эффективная стратегия фармакотерапии БП.
Современная стратегия терапии БП предусматривает применение средств патогенетического воздействия при одновременной профилактике лекарственных побочных явлений, возникающих ввиду значительной токсичности многих препаратов при многолетнем назначении [1, 23, 26].
Г.Н. Крыжановским и соавт. (2002) сформулирована концепция комплексной патогенетической терапии (КПТ) болезни Паркинсона. Принцип КПТ заключается в сочетанном воздействии лечебных средств на разные звенья патологического процесса. Поскольку между звеньями патологической системы паркинсонического синдрома существуют взаимопотенциирующие связи, такая терапия дает более значительный лечебный эффект по сравнению с результатом действия каждого препарата на отдельное звено патологической системы (монотерапия). Этот эффект может быть достигнут применением антипаркинсонических средств в уменьшенных дозах, что снижает вероятность потенциации нейротоксического действия препаратов и развития побочных проявлений с вторичными лекарственными синдромами, борьба с которыми может быть труднее, чем с исходной болезнью.
Основные направления комплексной патогенетической терапии должны сводиться к следующему:
— регуляция дофаминергической трансмиссии в функциональных условиях ДА-синапса и пула ДА-нейронов;
— управление синтезом ДА путем воздействия на недофаминергическую нейротрансмиссию;
— уменьшение степени прогрессирования заболевания с помощью нейропротекции поврежденных ДА-нейронов.
Поскольку дефицит ДА в нигростриатуме при БП является ключевым механизмом патогенеза заболевания, больные нуждаются в дофаминергической заместительной терапии. Общепризнано, что леводопа, т.е. левовращающий изомер ДА, проникающий через гематоэнцефалический барьер (ГЭБ), является наиболее адекватным препаратом для контроля паркинсонических симптомов. В настоящее время леводопа в чистом виде не применяется ввиду выраженных побочных эффектов. Категорически нельзя считать оправданными прежние рекомендации доводить суточную дозу препарата до 3–8 г. Современные леводопасодержащие препараты состоят из комбинации леводопы с ингибитором дофадекарбоксилазы карбидопой или бенсеразидом. Эти ингибиторы не проникают через ГЭБ и блокируют превращение экзогенно вводимой леводопы в ДА только на периферии, увеличивая тем самым концентрацию медиатора в нигростиратуме. Цель леводопа-терапии — не ликвидировать полностью все симптомы заболевания, а в достаточной степени улучшить состояние больного при как можно меньшей суточной дозе [21, 22].
По силе влияния на симптомы паркинсонизма леводопасодержащие препараты превосходят все остальные средства, имеющиеся в распоряжении современной фармакотерапии этого заболевания. На ранних стадиях заболевания у некоторых больных леводопа может полностью купировать все симптомы, делая больного внешне здоровым. Этот период лечения в специальной литературе носит образное название honeymoon — «медовый месяц» [2].
По мнению большинства специалистов, лечение леводопой, являясь классической заместительной терапией, вызывает улучшение состояния больных, но не останавливает прогрессирование заболевания [21, 26].
Две основные причины — способность леводопы быть нейротоксичной и появление леводопавызванных осложнений при продолжительном лечении — заставляют воздерживаться от заместительной терапии до тех пор, пока паркинсонические симптомы явно не станут инвалидизировать больных.
Другие средства, в том числе дофаминергические (амантадин, агонисты ДА-рецепторов, селегилин и др.), не могут конкурировать с леводопасодержащими препаратами по выраженности антипаркинсонического действия.
На ранних стадиях БП в качестве терапии первого этапа целесообразно применять ДА-агонисты, которые в современных условиях играют все большую роль и в лечении выраженных стадий БП с целью леводопа-экономного эффекта, особенно на фоне уже развившихся осложнений лечения. Кроме того, ДА-агонисты стимулируют пресинаптические рецепторы, что снижает уровень окислительного стресса и обеспечивает нейропротекторный эффект в отношении ДА-нейронов [24, 27, 29].
Важным направлением в патогенетической терапии БП является применение ингибиторов катаболизма ДА в базальных ганглиях. Считается, что ингибиторы МАО-Б должны назначаться больным БП в качестве препаратов безусловного выбора сразу же, как только диагностировано заболевание. На начальных стадиях ингибиторы МАО-Б можно применять в виде монотерапии, на более поздних — в сочетании с леводопасодержащими препаратами, что снижает дозировки последних на 20–30 % и уменьшает побочные явления многолетней лекарственной терапии [26].
К новой генерации противопаркинсонических препаратов относятся ингибиторы КОМТ-фермента, участвующего в распаде ДА [1, 2, 8, 23]. Такой препарат, как энтакапон, зарекомендовал себя как облигатное средство, предназначенное для лечения и профилактики вызванных леводопой гиперкинезов и дискинезий. В настоящее время синтезирован и применяется в странах Европы препарат сталево, являющийся комбинацией леводопы/карбидопы с энтакапоном в одной таблетке.
Отдельной оценки заслуживает многолетняя практика применения антихолинергических средств. Считается, что клиническое применение антихолинергиков должно быть строго дифференцированным ввиду частого развития таких побочных эффектов, как ослабление когнитивных функций, деменция, галлюцинации, тазовые расстройства. Полагают, что это может быть связано со специфическим эффектом воздействия данного класса препаратов на холинергические нейроны коры, особенно у больных БП с повышенным риском развития деменции. По данным статистики, у больных, не получавших антихолинергики, клинические симптомы деменции возникают в 46 % случаев, а у пациентов, принимавших эти препараты, — в 93 % случаев [8, 26]. Отдавая должное важности указанных фактов для клиницистов, следует подчеркнуть, что антихолинергики по-прежнему имеют большое значение в патогенетической терапии БП, особенно в случаях недостаточной эффективности препаратов дофаминергического ряда.
Современные успехи в лечении БП связаны с новыми представлениями о механизме действия амантадина. Эти препараты стимулируют высвобождение ДА из нейрональных депо, повышают чувствительность рецепторов к дофамину, тормозят процесс обратного поглощения медиатора пресинаптическим нейроном [16]. Показано, что амантадин значительно ослабляет возбуждающие глутаматные кортико-стриатные влияния на холинергические нейроны, ввиду чего препараты этого класса относят к группе антагонистов глутаматных рецепторов, к непрямым антихолинергикам и нейропротекторам. В качестве самостоятельного средства амантадин хорошо зарекомендовал себя в лечении начальных стадий БП, а применение его в комплексной терапии заболевания позволяет уменьшить суточные дозы леводопы и выраженность побочных явлений [30].
К новым технологиям лекарственной терапии следует отнести дуодопа-помпу, апоморфин в помпе и для подкожных инъекций, новый селективный ингибитор МАО-Б разагилин (азилект) и агонист ДА-рецепторов ротиготин в пластыре, показания к назначениям которых должны быть рассмотрены отдельно.
Представленные классы лекарственных средств патогенетического действия эффективно влияют на клиническую симптоматику БП. Однако происходящая при этом заболевании необратимая дегенерация значительной популяции ДА-нейронов нигростиатума определяет невозможность полного излечения и обосновывает необходимость пожизненного приема противопаркинсонических препаратов.
Как уже было отмечено, одним из стратегических направлений консервативного лечения БП является использование принципа КПТ, который заключается в сочетанном медикаментозном воздействии на отдельные звенья патогенеза заболевания с целью взаимного потенциирования эффектов препаратов и минимализации их суточных дозировок [1].
В процессе длительного (в течение 8–10 лет) наблюдения за клиническим течением БП у 276 больных, находящихся на учете в отделении экстрапирамидных заболеваний нервной системы ГУ «Институт геронтологии им. Д.Ф. Чеботарева НАМН Украины», были проанализированы особенности влияния КПТ на основные симптомы заболевания и частоту побочных явлений при применении основных классов противопаркинсонических препаратов [3, 6].
Этапность лечения БП определялась хроническим прогрессирующим характером течения заболевания и стадийностью патологического процесса. Подбор фармакологических средств в терапевтический комплекс был обусловлен стадией заболевания, доминирующим клиническим синдромом, индивидуальной переносимостью препаратов и зависел от психосоматического статуса, возраста больного и длительности заболевания. Однако, несмотря на соблюдение этих правил, у большинства больных неизбежно возникали побочные эффекты при многолетней фармакотерапии БП, что в значительной степени затрудняло возможность эффективного патогенетического лечения.
Установлено, что наиболее часто побочные эффекты наблюдались при применении дофасодержащих средств (ДСС) — 58 % случаев; несколько реже — при назначении амантадина (44 %) и холинолитиков (31 %). Реже всего побочные действия отмечались при лечении ингибиторами МАО-Б (9 %). При лечении холинолитиками — побочные эффекты со стороны вегетативной нервной системы (15 %) в виде сухости во рту, нарушения аккомодации, мочеиспускания. Терапия амантадином почти одинаково часто вызывала вегетативные побочные эффекты (13 %), а также неврологические (11 %) в виде возбуждения, бессонницы, головокружения, головной боли. Терапия дофасодержащими средствами чаще всего осложнялась неврологическими побочными эффектами (41 %), большинство из которых составляли лекарственные дискинезии.
Частота побочных эффектов препаратов леводопы в значительной степени определяется ее средней суточной дозой. Анализ полученных результатов показал, что у больных, принимавших леводопу с ингибиторами дофадекарбоксилазы (ИДК) при суточной дозе леводопы до 750 мг, частота гастроэнтеральных, сердечно-сосудистых и вегетативных побочных эффектов достаточно низкая. В то же время частота неврологических побочных эффектов при приеме максимальной дозы достигала 58 %. Если больной принимал ДСС в дозе, превышающей эмпирически установленную субмаксимальную суточную дозу (500 мг в пересчете на леводопу), частота побочных явлений значительно возрастала по сравнению с приемом дозы 375–500 мг: частота гастроэнтеральных побочных явлений — в 7 раз, сердечно-сосудистых — в 3–4 раза, психических — в 4 раза и неврологических — в 2 раза. Этот факт является дополнительным подтверждением того, что многолетнее лечение БП с помощью препаратов леводопы/ИДК следует проводить, не превышая субмаксимальную дозу леводопы 500 мг [3, 6].
При длительном течении БП и многолетней терапии ДСС наступают изменения клинической картины заболевания, так называемый клинический патоморфоз симптомов, к основным проявлениям которого относятся дискинезии, а также ряд специфических феноменов, проявляющихся колебаниями двигательной активности в виде моторных флуктуаций в течение суток [1, 2, 8, 26]. К этим феноменам относятся: феномен истощения эффекта разовой дозы (wearing-off — «изнашивание»), феномен «включение-выключение» (on-off), феномен «застывание» (freezing). Разнообразные дискинезии включают: дистонию действия и периода «выключения», дискинезию «пика дозы», двухфазную дискинезию, пароксизмальную непредсказуемую дискинезию и др. У больных БП, получавших леводопу/ИДК свыше 5 лет, наиболее часто (в 48 % случаев) наблюдается дискинезия «пика дозы»; каждый из других вариантов наблюдался с частотой от 8 до 22 %.
Частота лекарственных дискинезий, истощения эффекта суточной дозы ДСС, неравномерность действия ДСС в течение суток, феноменов «включение-выключение» и «застывание» нарастают при увеличении продолжительности болезни. Выраженность феномена истощения эффекта суточной дозы ДСС, совпадающая со снижением эффективности лечения, значительно возрастает после 10 лет течения заболевания [3, 6, 8].
Частота всех типов клинического патоморфоза увеличивается и при нарастании степени тяжести БП. Такие проявления клинического патоморфоза, как неравномерность действия ДСС в течение суток, когда одна и та же однократная доза ДСС хуже действует в определенные часы после приема, а также феномены «включение-выключение» и «застывание» наблюдались только у больных БП при 3-й и 4-й стадиях заболевания.
Следует подчеркнуть, что проявления клинического патоморфоза симптомов БП нарастали у больных при увеличении суточной дозы ДСС, особенно при увеличении эмпирической максимальной суточной дозы леводопы выше 750 мг.
При анализе возрастных критериев риска или преимуществ использования основных антипаркинсонических средств, в том числе леводопасодержащих препаратов, мы считаем приемлемым рубежом для оценки эффективности их действия условные периоды до и после 60 лет. Поскольку лечение леводопасодержащими препаратами у пациентов моложе 60 лет с ранним началом заболевания (до 45 лет) часто осложняется развитием моторных флуктуаций и непроизвольных движений, следует считать проблематичной возможность долговременного применения препаратов в этой группе больных, а суточная доза леводопы не должна превышать 200–400 мг до тех пор, пока не появятся клинические признаки прогресса заболевания [2, 6].
Побочные явления в результате длительной леводопа-терапии возникали сравнительно редко у больных старше 60 лет. По результатам долговременного наблюдения за 156 пациентами с БП среднего (49–59 лет) и пожилого (60–74 года) возраста с различной выраженностью заболевания, в течение 10–15 лет получавших леводопу/ИДК, установлено, что эффективность лечения зависит от возраста больного к началу болезни и длительности лечения. Так, эффективность леводопа-терапии снижается при увеличении приема препаратов, что более выражено у больных среднего возраста. Значительная эффективность лечения в начале болезни чаще сочетается с возникновением побочных явлений впоследствии. Дискинетический синдром как осложнение леводопа-терапии более выражен у больных среднего возраста; в этом же возрасте он с наибольшей частотой трансформируется в феномен «включение-выключение». Когнитивные дисфункции у больных пожилого возраста как возможное лекарственное осложнение могут быть обусловлены и многолетним применением препаратов леводопы.
В структуру КПТ при БП входит и нейрохирургическое лечение [31]. Считают, что применение нейрохирургических методов лечения БП оправдано в случаях явной неэффективности консервативной терапии. Показаниями к хирургическому методу лечения являются значительное снижение эффективности леводопа-терапии, прогрессирующая инвалидизация. Показания к операции следует определять с непременным учетом этико-деонтологических аспектов. Больные и члены их семей должны быть информированы об отсутствии абсолютных гарантий пользы хирургического лечения, вероятности развития постоперационных осложнений с риском для жизни в 1–2 % случаев, необходимости продолжать пожизненный прием патогенетической терапии, так как операция не избавляет больного от БП, а лишь смягчает отдельные симптомы заболевания.
Последние полтора десятка лет охарактеризовались внедрением в практику лечения болезней движений принципиально новой функциональной нейрохирургической технологии – высокочастотной электростимуляции глубинных отделов мозга, или глубинной стимуляции мозга (ГСМ), в англоязычной литературе — deep brain stimulation. Она была впервые предложена группой французских нейрохирургов из Гренобля (A.L. Benabid, P. Pollak). Сущность метода заключается в стереотаксической имплантации электрода в мозговую «мишень» и осуществлении его стимуляции импульсным генератором в специально подобранном режиме. Данная технология прошла успешную апробацию на тысячах больных с паркинсонизмом (особенно при дрожательном фенотипе заболевания), различными формами идиопатической и симптоматической дистонии, эссенциальным тремором и другими тяжелыми и нередко некурабельными состояниями. Технология ГСМ доказала свою высокую эффективность и надежность, низкий риск побочных эффектов, возможность осуществления долговременной стимуляции с поддержанием достойного качества жизни оперируемых пациентов и членов их семей. Важным преимуществом ГСМ является возможность проведения двусторонней операции без опасности развития нарушений бульбарных функций [32]. В то же время следует отметить, что, несмотря на несомненную клиническую эффективность операции, после проведения ГСМ не достигается прекращения прогрессирования нейродегенеративного процесса. Обращает на себя внимание также появление сообщений, касающихся некоторых осложнений со стороны эмоционально-волевой, поведенческой и когнитивной сфер при хронической многолетней ГСМ, что требует детального дальнейшего исследования и анализа тонких нейрофизиологических механизмов действия данной процедуры.
Украинская ассоциация по проблеме болезни Паркинсона
Специалисты украинской школы, занимающиеся проблемами паркинсонизма, прошли значительный путь, ознаменовавшийся немалыми достижениями. Научные и практические разработки в области диагностики и лечения этого заболевания позволили еще в 1972 году (по инициативе профессора Н.Б. Маньковского) открыть на базе Института геронтологии АМН СССР (в настоящее время — ГУ «Институт геронтологии им. Д.Ф. Чеботарева НАМН Украины») первое в странах постсоветского пространства специализированное отделение по экстрапирамидным заболеваниям с центром паркинсонизма. Избранное направление и методы оказания медицинской помощи больным паркинсонизмом оказались весьма перспективными для дальнейшего развития.
Очередным организационным этапом создания структуры общественной медицинской организации, объединяющей больных БП и специалистов, занимающихся проблемой паркинсонизма, было создание в 1994 году Украинской ассоциации по проблеме паркинсонизма. Ассоциация подняла работу по социальной реабилитации больных на более высокий уровень.
В 1997 году наша Ассоциация принята в Европейский паркинсонический союз (EPDA). В 2001, 2004 и 2009 гг. в Киеве по инициативе EPDA проведены международные симпозиумы «Болезнь Паркинсона: реальность и перспективы», на которых были обсуждены современные аспекты диагностики, лечения и реабилитации больных БП. Следует полагать, что перспективными направлениями терапии будущего являются разработка новых и совершенствование современных лекарственных форм и методов стереотаксической нейрохирургии, активация синтеза эндогенных нейротрофических факторов, а также имплантация клеток, преформированных на синтез дофамина и трофогенов методами генной инженерии.
1. Болезнь Паркинсона (этиология, патогенез, клиника, диагностика, лечение, профилактика) / Г.Н. Крыжановский, И.Н. Карабань, С.В. Магаева, В.Г. Кучеряну, Н.В. Карабань. — М.: Медицина, 2002. — 335 с.
2. Голубев В.Л., Левин Я.И., Вейн А.М. Болезнь Паркинсона и синдром паркинсонизма. — М.: Медпресс, 1999. — 416 с.
3. Карабань Н.В. Комплексна патогенетична терапія хвороби Паркінсона (клінічні, діагностичні, медико-соціальні аспекти): Автореф. дис... д-ра мед. наук. — К., 2007. — 33 с.
4. Левин О.С. Психические расстройства при болезни Паркинсона и их коррекция // Экстрапирамидные расстройства. Руководство по диагностике и лечению / Ред. В.Н. Шток, И.А. Иванова-Смоленская, О.С. Левин. — М.: Медпресс-информ, 2002. — С. 125-151.
5. Маньковский Н.Б., Вайншток А.Б., Олейник Л.И. Сосудистый паркинсонизм. — К.: Здоров’я, 1982. — 208 с.
6. Маньковский Н.Б., Карабань Н.В. Особенности клинического течения и фармакотерапии болезни Паркинсона на разных этапах развития заболевания // Междунар. мед. журнал. — 2005. — Т. 11, № 4. — С. 47-51.
7. Фролькис В.В. Старение и увеличение продолжительности жизни. — Л.: Наука, 1988. — 239 с.
8. Шток В.Н., Федорова Н.В. Болезнь Паркинсона // Экстрапирамидные расстройства. Руководство по диагностике и лечению / Ред. В.Н. Шток, И.А. Иванова-Смоленская, О.С. Левин. — М.: Медпресс-информ, 2002. — С. 87-124.
9. Яхно Н.Н., Захаров В.В., Локшина А.Б., Коберская Н.Н., Мхитарян Э.А. Деменция: руководство для врачей. — М.: Медпресс-информ, 2010. — 264 с.
10. Braak H., Del Tredici K., Rub V. et al. Staging of brain pathology related to sporadie Parkinson’s disease // Neurobiol. Aging. — 2003. — 24. — P. 194-211.
11. Brooks D.J. The early diagnosis of Parkinson’s disease // Ann. Neurol. — 1998. — 44 (suppl. 1). — P. 10-18.
12. Brooks D.J. Monitoring neuroprotection and restorative therapies in Parkinson’s disease with PET // J. Neural. Transm. Suppl. — 2000. — 60. — P. 125-137.
13. Fahn S., Elton R.L. For the UPDRS Committee. Unified Parkinson’s Disease Rating Scale // Recent Developments in Parkinson’s disease / Eds S. Fahn, C.D. Marsden, D. Calne, M. Goldstein. — Florham Park, NJ: McMillan Healthcare Information, 1987. — P. 153-163.
14. Hartmann A., Agid Y., Schapira A. Pathophysiology of Parkinson’s disease // Parkinsonian Disorders in Clinical Practice / Eds. A. Schapira, A. Hartmann, Y. Agid. — Blackwell Publishing Ltd, 2009. — P. 1-10.
15. Hughes A.J., Daniel S.E., Kilford L., Lees A.J. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases // J. Neurol. Neurosurg. Psychiatry. — 1992. — 55. — P. 181-184.
16. Inzelberg R., Bonuccelli V., Schechtman E. Association between amantadine and the onset of dementia in Parkinson’s dise-ase // Mov. Disord. — 2006. — 21. — P. 1375-1379.
17. Jellinger K.A. Lewy body-related alpha-synucleinopathy in the aged human brain // J. Neural. Transm. — 2004. — 111. — P. 1219-1235.
18. Jenner P. Factors influencing the onset and persistence of dyskinesia in MPTP treated primates // Ann. Neurol. — 2000. — 47 (4 suppl.1). — P. 90-99.
19. Langston J.W. The Parkinson’s complex: parkinsonism is just the tip of the iceberg // Ann. Neurol. — 2006. — 59. — P. 591-596.
20. De Lau L.M., Breteler M.M. Epidemiology of Parkinson’s disease // Lancet Neurol. — 2006. — 5. — P. 525-535.
21. Nutt J.G. Pharmacokinetics and pharmacodynamics of levodopa // Mov. Disord. — 2008. — 23 (suppl. 3). — P. S580-S584.
22. Obeso J.A., Olanow C.W., Nutt J.G. Levodopa motor complications in Parkinson’s disease // Trends Neurosci. — 2000. — 23 (suppl.10). — P. S2-S7.
23. Olanow C., Watts R., Koller W. An algorithm (decision tree) for the management of Parkinson’s disease (2001): treatment guidelines // Neurology. — 2001. — V. 56 (suppl. 5). — P. 1-88.
24. Olanow C.W., Schapira A.H., Agid Y. Neuroprotection for Parkinson’s disease: prospects and promises // Ann. Neurol. — 2003. — 53 (suppl. 3). — P. S1-S2.
25. Olanow C.W. Pathogenesis of cell death in Parkinson’s dise-ase // Mov. Disord. — 2007. — 22. — P. S335-S342.
26. Olanow C.W., Stern M.B., Sethi K. The Scientific and Clinical Basis for the Treatment of Parkinson’s Disease // Neurology. — 2009. — V. 72, № 21 (suppl. 4). — P. S1-S136.
27. Pfeiffer R.F., Gutmann L., Hull L. et al. Continued efficacy and safety of subcutaneous apomorphine in patients with advanced Parkinson’s disease // Park. Relat. Disord. — 2007. — 13. — P. 93-100.
28. Stacy M.A., Street V. Dopamine Agonists // Handbook of Parkinson’s disease. Fourth edition / Eds R. Pahwa, K.E. Lyons. — NY; London; Informa: Healthcare, 2007. — P. 335-338.
29. Schapira A., Olanow C.W. Neuroprotection in Parkinson’s disease: myths, mysteries and misconceptions // JAMA. — 2004. — 291. — P. 358-364.
30. Schapira A.H. The clinical relevance of levodopa toxicity in the treatment of Parkinson’s disease // Mov. Disord. — 2008. — 23 (suppl. 3). — P. S515-S520.
31. Walter B.L., Vitek J.L. Surgical treatment for Parkinson’s disease // Lancet Neurol. — 2004. — 3. — P. 719-728.
32. Weaver F.M., Tollett K., Stein M. et al. Bilateral deep brain stimulation vs best medical therapy for patients with advanced Parkinson’s disease: a randomized controlled trial // JAMA. — 2009. — 301. — P. 63-73.