
Международный неврологический журнал 7 (45) 2011
Вернуться к номеру
Окулофарингеальная миодистрофия. Научный обзор и описание клинического случая поздней диагностики заболевания у взрослого пациента
Авторы: Евтушенко С.К., Иванова М.Ф., Шаймурзин М.Р., Симонян В.А., Марусиченко Е.А., Палагута А.В., Донецкий национальный медицинский университет им. М. Горького, Клиника ангионеврологии ГУ «Институт неотложной и восстановительной хирургии им. В.К. Гусака НАМН Украины», г. Донецк
Рубрики: Неврология
Версия для печати
В научном обзоре представлены особенности клинико-инструментальной диагностики окулофарингеальной миодистрофии в зависимости от типа наследования, степени ограничения двигательной функции, виды темпов прогрессирования. Описан клинический случай поздней диагностики окулофарингеальной миодистрофии у взрослого пациента.
Нейромышечные заболевания, окулофарингеальная мышечная дистрофия.
Окулофарингеальная мышечная дистрофия (ОФМД) относится к прогрессирующим аутосомным мышечным дистрофиям из группы нейромышечных заболеваний (НМЗ). Нейромышечные заболевания — клинически и генетически гетерогенная группа наследственно-дегенеративных заболеваний, характеризующихся прогрессирующей мышечной слабостью и атрофиями. В 2005 г. в Украине было зарегистрировано 1540 детей, страдающих нейромышечной патологией [3]. Ежегодно выявляется около 200 первичных больных, и, к сожалению, смертность среди этой категории пациентов остается высокой.
Согласно современной классификации НМЗ [1–3, 10], выделяют основные их формы:
1. Прогрессирующие мышечные дистрофии:
1.1. Х-сцепленные мышечные дистрофии.
1.2. Аутосомные мышечные дистрофии (сюда относят ОФМД).
2. Врожденные миодистрофии.
3. Спинальные амиотрофии:
3.1. Проксимальные спинальные амиотрофии детского возраста.
3.2. Редкие формы спинальных амиотрофий в детском возрасте.
3.3. Спинальные амиотрофии взрослых.
4. Наследственные нейропатии, сопровождающиеся амиотрофиями:
4.1. Наследственные мотосенсорные нейропатии.
4.2. Наследственные сенсорные нейропатии.
4.3. Варианты наследственных мотосенсорных нейропатий.
5. Врожденные структурные миопатии:
5.1. Болезнь центрального стержня (MIM 117 000).
5.2. Болезнь множественных центральных стержней.
5.3. Врожденная миопатия с диспропорцией типов волокон.
5.4. Миотубулярная (центронуклеарная) миопатия (MIM 310 400).
5.5. Немалиновая (палочковидная) миопатия.
5.6. Врожденная миопатия с внутрицитоплазматическими включениями в виде редуцированных телец.
5.7. Миопатия с накоплением телец, сходных с отпечатками пальцев.
5.8. Саркотубулярная миопатия.
6. Синдром ригидного позвоночника.
7. Множественный врожденный артрогриппоз.
8. Метаболические миопатии:
8.1. Миопатические синдромы при гликогенозах.
8.2. Митохондриальные болезни, обусловленные мутациями митохондриальной ДНК.
8.3. Митохондриальные болезни, обусловленные мутациями ядерной ДНК.
9. Воспалительные миопатии:
9.1. Полимиозит.
9.2. Дерматомиозит.
9.3. Острый инфекционный миозит.
9.4. Миозит с включениями телец.
9.5. Х-сцепленная вакуольная миопатия.
9.6. Гранулематозный миозит.
10. Наследственные миотонические синдромы и пароксизмальные миоплегии:
10.1. Стационарные, медленнопрогрессирующие миотонии.
10.2. Периодические (рецидивирующие) формы миотонии.
10.3. Наследственные пароксизмальные миоплегии.
На первом десятилетии жизни дебютирует большинство клинических форм прогрессирующих мышечных дистрофий (ПМД) и амиотрофий, имеющих прогредиентное течение и на сегодняшний день неизлечимых. Вместе с тем в последнее десятилетие достигнуты значительные успехи в изучении молекулярных механизмов наследственных нейромышечных заболеваний, разрабатываются пути внутриутробной ДНК-диагностики. Практически при всех формах нейромышечной патологии выявлены конкретные мутантные гены на хромосомах. Согласно клинико-молекулярной генетической классификации [26], опубликованной в 9-м номере журнала Neuromuscular Disorders (1999), в настоящее время можно выделить дистрофинопатии, эмеринопатии, ламинопатии, конечностно-поясные мышечные дистрофии, дистальные типы, окулофарингеальные типы, врожденные мышечные дистрофии [2, 10]. В «Международном неврологическом журнале» (№ 4(14), 2007) можно ознакомиться с полной адаптированной [3] к использованию в клинической практике клинико-молекулярной классификацией нейромышечных заболеваний, где авторы указали MIM-коды их различных форм.
MIM-коды — менделевское наследование (Mendelian Inheritance in Man) — это результат исследований, проводимых на протяжении 30 лет в Университете Джона Хопкинса в Балтиморе под руководством профессора Виктора Мак-Кьюсика и позволяющих систематизировать информацию о генетических картах хромосом человека, локализации и функциях отдельных генов и структуре генома в целом. С 2-годичным интервалом издаются обновляемые энциклопедии, содержащие сводные данные о картированных генах человека и связанных с ними наследственных болезнях под названием «Менделевское наследование у человека: каталог человеческих генов и генетических болезней» (Mendelian inheritance in man. Catalogs of autosomal dominant, autosomal recessive, and X-linked phenotypes). Так, после названия формы генетически детерминированного нейромышечного заболевания указывается его международный шестизначный номер (MIM), в котором первое число означает тип наследственности. Издания каталога В. Мак-Кьюсика выпускаются как в печатной форме, так и в электронном варианте (OMIM — Online Mendelian Inheritance in Man) [1, 6].
Дистрофия (древнегреч. dystrophe, от dys… — приставка, означающая затруднение, нарушение, и trophe — питание) — патологический процесс, в результате которого та или иная ткань теряет или накапливает вещества, в норме не характерные для нее. Дистрофия характеризуется повреждением клеток и межклеточного вещества, в результате чего изменяется функция органа. В основе дистрофии лежит нарушение трофики, то есть комплекса механизмов, обеспечивающих метаболизм и сохранность структуры клеток и тканей. Трофические механизмы делят на клеточные и внеклеточные. Клеточные механизмы обеспечиваются самой структурой клетки и ее саморегуляцией, благодаря чему каждая клетка осуществляет свойственную ей функцию. Внеклеточные механизмы включают в себя систему транспорта продуктов метаболизма (кровяное и лимфатическое микроциркуляторное русло), систему межклеточных структур мезенхимального происхождения и систему нейроэндокринной регуляции обмена веществ. При нарушении в любом звене механизмов трофики может возникнуть тот или иной вид дистрофии.
По виду нарушения обменных процессов выделяют следующие дистрофии: белковую, жировую, углеводную, минеральную, водную.
Миопатии (греч. mys, myos — мышца + pathos — страдание, болезнь) — это группа гетерогенных нервно-мышечных заболеваний, характеризующихся прогрессирующим развитием первичного дистрофического или вторичного (денервационного: спинального или неврального) атрофического процесса в скелетной мускулатуре, сопровождающегося мышечной слабостью и двигательными нарушениями.
К миопатиям относят как наследственные нервно-мышечные заболевания (отличающиеся возрастом дебюта заболевания, тяжестью течения и преимущественным поражением тех или иных групп мышц), так и разнообразные нервно-мышечные синдромы при ряде соматических и неврологических болезней [4].
В эту группу заболеваний входят:
1. Мышечные дистрофии.
2. Врожденные миопатии.
3. Воспалительные миопатии.
4. Метаболические миопатии.
5. Токсические миопатии.
В МКБ-10 заболевания мышц представлены в рубриках: G71 («Первичные поражения мышц», исключены: артрогриппоз множественный врожденный (Q74.3), нарушения обмена веществ (Е70-Е90), миозит (М60.-), G72 («Другие миопатии», исключены: врожденный множественный артрогриппоз (Q74.3), дерматополимиозит (МЗЗ.-), ишемический инфаркт мышцы (М62.2), миозит (М60.-), полимиозит (МЗЗ.2)), С73* («Поражения нервно-мышечного синапса и мышц при болезнях, классифицированных в других рубриках»).
Прогрессирующие мышечные дистрофии (ПМД) — группа наследственных заболеваний, вызывающих дегенерацию мышечной ткани и проявляющихся прогрессирующей слабостью скелетных мышц. Выделяют следующие варианты мышечных дистрофий, различающихся генетическим дефектом, типом наследования, возрастом появления, темпом прогрессирования, вовлечением определенных групп мышц. В зависимости от преимущественной топографии мышечного поражения выделяют следующие ПМД: дистальные, проксимальные, поясно-конечностные, окулярные и окулофарингеальные.
Каждая из этих групп включает различное число генетически гетерогенных вариантов. Описаны нозологические формы ПМД с аутосомно-доминантным, аутосомно-рецессивным и Х-сцепленным рецессивным типами наследования. В рамках одного генетического варианта выделяются аллельные серии, обусловленные различными мутациями в одном и том же гене.
Характерными клиническими признаками ПМД являются симптомы вялого паралича в различных группах мышц без признаков поражения мотонейронов и периферических нервов.
На электромиограмме выявляется типичный первично-мышечный паттерн, характеризующийся снижением амплитуды М-ответа, усилением интерференции и полифазности потенциала.
Морфологический дефект, выявляемый в биоптате мышечного волокна, характеризуется атрофией, жировым перерождением и некрозом мышечных волокон с наличием их регенерации, а также разрастанием соединительной ткани эндомизия. При некоторых нозологических формах выявляются специфичные для врожденных доброкачественных структурных миопатий изменения мышечных волокон, такие как центральное расположение ядер или наличие обрамленных вакуолей.
Точная диагностика отдельных нозологических форм возможна только при проведении молекулярно-генетического анализа, направленного на выявление мутаций в том или ином гене, и в ряде случаев — исследовании концентрации того или иного белка в биоптате мышечного волокна.
При установлении диагноза в развернутой формулировке указываются стадия, преимущественно вовлекаемые мышцы, степень ограничения двигательной функции, выраженность сопутствующих проявлений (умственная отсталость, кардиомиопатия), темп прогрессирования.
Выделяют три стадии мышечной дистрофии:
I стадия — умеренно выраженные двигательные нарушения (слабость выявляется лишь при значительной нагрузке);
II стадия — выраженные двигательные затруднения при ходьбе, подъеме по лестнице, выполнении физической работы;
III стадия — параличи, грубые контрактуры, самостоятельное передвижение невозможно.
Варианты течения мышечных дистрофий (темпы прогрессирования):
1) быстрое прогрессирование — способность к самостоятельному передвижению утрачивается через 5 лет от начала болезни;
2) средний темп прогрессирования — утрата способности к самостоятельному передвижению через 10 лет от начала болезни;
3) медленное прогрессирование — отсутствие выраженных двигательных нарушений через 10 лет от начала болезни.
Степень выраженности двигательного дефекта:
1) легкая — затруднено выполнение сложных движений, нагрузочных тестов, слабость при длительной физической нагрузке (более 2–3 ч), легкие ограниченные амиотрофии, сила мышц — 4 балла;
2) умеренная — затруднено выполнение обычных движений, необходим отдых после непродолжительной нагрузки (1–2 ч), умеренная атрофия больших групп мышц, сила мышц — 3 балла;
3) выраженная — крайне затруднены обычные движения, однако передвижение и самообслуживание возможны, хотя и в ограниченном объеме; значительно выражены генерализованные мышечные атрофии, сила мышц — 2 балла;
4) значительно выраженная — распространенные мышечные атрофии, выраженные нарушения функции мышц конечностей и туловища, дыхательных мышц, сила мышц — 1 балл, контрактуры многих суставов, самостоятельное передвижение невозможно, в быту — полная зависимость от окружающих.
Окулофарингеальная мышечная дистрофия — заболевание, которое встречается в двух генетических вариантах: аутосомно-рецессивном (OMIM 257 950) и аутосомно-доминантном (OMIM 164 300).
Эти варианты болезни являются аллельными и обусловлены различными мутациями в одном гене.
Клинические варианты описаны в зависимости от типа наследования.
1. Аутосомно-доминантный вариант заболевания впервые описан Victor и соавтор. в 1962 году у 9 членов одной семьи из трех поколений [6] . Первые симптомы возникали на 4–5-м десятилетиях жизни и в большинстве случаев характеризовались сочетанием дисфагии с прогрессирующим птозом верхних век. По мере прогрессирования заболевания отмечалось распространение симптомов мышечной слабости на мышцы плечевого и тазового поясов. Satoyoshi & Kinoshita [8] в 1977 году описали семью с аутосомно-доминантной сегрегацией окулофарингеальной миопатии, характеризующейся значительной генерализацией процесса по мере течения болезни. У наблюдаемых больных мышечная слабость распространялась на мышцы лица, шеи, дистальных отделов конечностей, а также анального сфинктера. По мнению авторов, представленное ими наблюдение является отдельным вариантом болезни — окулофарингодистальным (OMIM 164 310). Это предположение вызвало сомнения, так как отмечено существование различной степени генерализации процесса у больных в одной и той же семье. Описаны единичные больные с наличием пигментной дегенерации сетчатки.
Большинство авторов [1, 9, 10, 12] эту форму аутосомно-доминантной миопатии относят к довольно редким, возникающим в зрелом возрасте и медленнотекущим заболеваниям, считают, что клинически болезнь проявляет себя как локальная миопатия. Поражаются мышцы, осуществляющие движения глазных яблок, и мышца, поднимающая верхнее веко. Фарингеальные расстройства обусловлены включением в процесс констрикторов глотки, что затрудняет глотание. Типична симметричность процесса. Начальные признаки болезни появляются в возрасте 30–40 лет: двустороннее опущение верхнего века при ограничении движения глазных яблок. Как правило, на диплопию больные не жалуются. Объяснение этому находят в медленном и симметричном развитии парезов глазодвигательных мышц. Значительно утяжеляют заболевание и ухудшают прогноз фарингеальные симптомы. Начинаясь с дисфагии, они имеют тенденцию к нарушению функций (афагии). Следует иметь в виду существование окулярной миопатии, при которой фарингеальные расстройства не выражены. Этот вариант миопатии рассматривается одними исследователями как самостоятельное заболевание, другими — как дебют окулофарингеальной миопатии.
2. Аутосомно-рецессивный вариант заболевания впервые описан Fried и соавт. в 1975 году у двух сестер, родившихся от кровнородственного брака. Для этой формы болезни характерно более раннее начало и вовлечение в процесс дистальных групп мышц конечностей [1, 7].
При электромиографическом обследовании больных с ОФМД определяют первично-мышечный характер поражения.
При морфологическом исследовании выявляются нитевидные образования в ядрах скелетных мышц. Эти нити имеют ветвящуюся трубчатую структуру и иногда поперечно исчерчены. Наряду с этим отмечаются атрофические изменения в мышечных волокнах 1-го типа. При электронном микроскопировании обнаруживается увеличение размеров митохондрий с наличием в них крестовидных включений. Также могут быть обнаружены вакуоли, при электронной микроскопии в них видны обрывки мембран, скопления гликогена и другие неспе-цифичные остатки лизосомного происхождения. Особенность окулофарингеальной миодистрофии — наличие в ядрах нитевидных трубочек диаметром 8,5 нм.
Аутосомно-доминантный и аутосомно-рецессивный варианты болезни обусловлены мутациями в одном и том же гене — РАВР2 (полиаденилсвязывающем протеине-2; OMIM 602 279), локализованном в области 14q11.2-q13. Основной тип мутаций — короткая экспансия тринуклеотидного повтора GCG в кодирующей части гена. В норме число повторов не превышает 6, однако у 2 % здоровых людей число повторов может достигать 7, что расценивается как проявление нормального полиморфизма. У больных с окулофарингеальной миопатией число повторов увеличено до 8–13. Тяжесть проявления заболевания зависит от количества повторов.
Антиципация, обусловленная увеличением количества повторов, не характерна. Возникновение аутосомно-рецессивного варианта обусловлено гомозиготностью по GCG7-повтору, который является примером аллели-модификатора. Наиболее тяжелый фенотип наблюдался у компаунд-гетерозигот GCG9/GCG7, а также гомозигот по GCG9-повторам. Патогенетически белок РАВР2 является высоконсервативным и содержится в ядре, где участвует в полиаденилировании мРНК. GCG-повторы кодируют включение полиаланинового тракта вблизи N-конца мутантного белка. Считается, что образующиеся в ядре нитевидные структуры представлены удлиненными нитями мутантного белка [1, 10, 11].
Заболевание особенно часто встречается у франко-язычных канадцев и в латиноамериканских семьях на юго-западе США. Оно описано также в большой еврейской семье восточноевропейского происхождения [1, 8, 9, 12].
На сегодняшний день для профилактики ОФМД возможна дородовая диагностика с использованием методов ДНК-анализа.
Несмотря на то что, согласно литературным данным, ОФМД относится к более поздним формам мышечных дистрофий, приводим клинический пример раннего дебюта ОФМД у пациента в возрасте 23 года, но установлен диагноз в 31 год.
Б. К., 31 год, был направлен в клинику неврологии Института неотложной и восстановительной хирургии им. В.К. Гусака НАМН Украины для получения консультативного заключения для МСЭК, но был госпитализирован для уточнения диагноза и находился в клинике с 5.04.2011 по 20.04.2011 г.
Обращали на себя внимание жалобы больного на периодическое покашливание при глотании, гнусавость голоса, опущение верхних век, выраженную общую слабость, исхудание.
Болеет с 23 лет (с 2005 года), когда впервые появились жалобы на гнусавость голоса, периодическую общую слабость. Обследовался и получал лечение на протяжении 3 месяцев по месту жительства у отоларинголога по поводу пансинусита, но без эффекта. Обследовался и лечился у невролога по месту жительства по поводу ВСД, астенического синдрома, рецидивирующего пансинусита, но состояние не изменялось. В течение 2 лет присоединилось опущение век в вечернее время, слабость в конечностях, покашливание при глотании. С 2007 года присоединились слабость в верхних и нижних конечностях. В апреле 2007 года обратился к отоларингологу областной клинической больницы г. Донецка с жалобами на гнусавость голоса, покашливания при глотании и исхудание. Рекомендована консультация невролога и торакального хирурга. Осмотрен торакальным хирургом, установлен диагноз: миастения, генерализованная форма, средней тяжести, стабильное течение, проба прозериновая — неубедительная. Госпитализирован в клинику неврологического отделения областной больницы. Обследован. ЛОР: без патологии, МРТ головного мозга: данных в пользу очаговой и объемной патологии не выявлено, расширены боковые желудочки и подоболочечные пространства; СКТ органов грудной клетки: данных в пользу гиперплазии вилочковой железы не выявлено. Установлен диагноз: вялотекущий энцефаломиелополирадикулоневрит с миастеническим и амиотрофическим синдромами (вялый, преимущественно дистальный тетрапарез). После проведенного лечения состояние не изменилось. С 2007 г. признан инвалидом 3-й группы по данному заболеванию. Продолжал прием калимина до 3 табл. в сутки, но состояние медленно ухудшалось. Прием калимина облегчения не приносил, сам отменил прием препарата. В связи с нарастанием мышечной слабости, исхуданием мышц, нарушением трудоспособности направлен в клинику неврологии ГУ «ИНВХ им. В.К. Гусака НАМН Украины» (г. Донецк).
Удалось уточнить: с детства состоял на диспансерном учете у невролога и хирурга по поводу плоскостопия и сколиоза. В армии не служил, комиссован по поводу заболевания опорно-двигательного аппарата (сколиоз).
Объективно: общее состояние относительно удовлетворительное. Астеничен, выражено общее исхудание, кожные покровы бледные, периферические лимфоузлы не увеличены. Следов немедикаментозных инъекций нет. В легких везикулярное дыхание, хрипов нет. Деятельность сердца ритмична, тоны приглушены, АД 100/60 мм рт.ст., пульс учащен до 90 уд/мин, ритмичен. Живот мягкий, безболезненный. Пульсация на периферических артериях стоп сохранена.
Неврологический статус: гипомимия, дисфония. Низкое стояние век, полуптоз с двух сторон, больше слева. Зрачки S ≥ D, фотореакции живые, парез отведения в обе стороны, парез взора вверх. Диплегия n.facialis. Мягкое небо неподвижно при фонации. Глоточный и небный рефлексы отсутствуют. Атрофий и фасцикуляций на языке нет. Фасцикуляций и фибрилляций на туловище также нет. Выраженная атрофия всех групп мышц туловища и конечностей. Сухожильные рефлексы рук отсутствуют, коленные угнетены, ахилловы отсутствуют. Тетрапарез преимущественно дистальный. Степпаж при ходьбе. Ретракция сухожилий. Расстройств чувствительности и координации нет (рис. 1–3: фото публикуются с информированного согласия пациента).
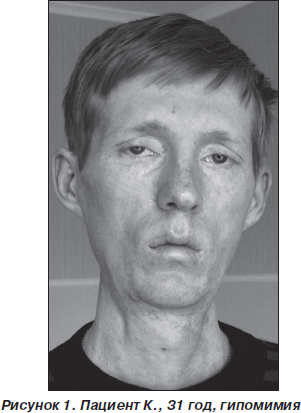
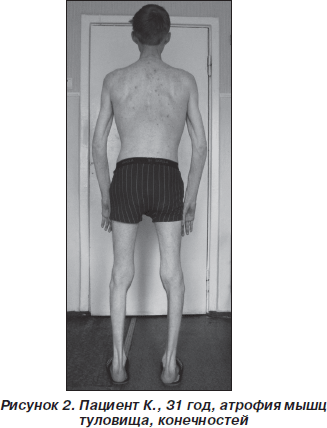
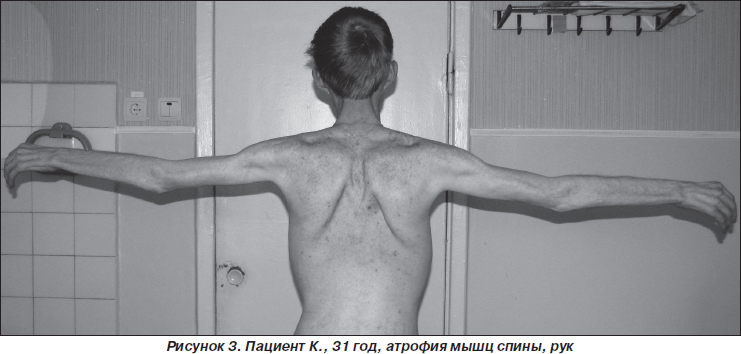
Результаты дообследования: общеклинические и биохимические анализы крови и мочи, в том числе на электролиты, сахар крови — без патологии, острофазовые реакции — в пределах нормы.
Креатинкиназа — 2821,90 Ед/л (норма 20,00–308,00 Ед/л), лактатдегидрогеназа — 364,48 Ед/л (норма 15,00–300,00 Ед/л).
Гормоны щитовидной железы — в пределах нормы.
УЗИ щитовидной железы: увеличенная, зернистая, неоднородная, эхогенность повышена, в правой доле мелкокистозные изменения до 4 мм в диаметре, явления хронического тиреоидита.
Эндокринолог: хронический тиреоидит, клинический эутиреоз.
Прозериновая проба: подкожно введено 2,0 мл прозерина, проба учитывалась около 40 мин, положительного эффекта не получено. Проба со льдом на веки — отрицательная.
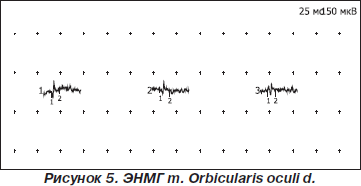
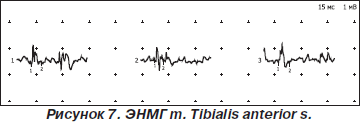
На электромиограмме: по данным игольчатой ЭМГ выявлены умеренные признаки первично-мышечного типа поражения двигательной единицы нейромышечного аппарата круговых мышц глаз, грудино-подъязычных, лопаточно-подъязычных, переднего брюшка двубрюшной мышцы, характеризующиеся уменьшением длительности до 48 % от нормы, снижением амплитуды потенциалов двигательной единицы до 45 % от нормы, нарастанием фазности потенциалов до 32 %, гистограмма распределения по длительности сдвинута влево.
Вместе с тем в мышцах дистальных отделов конечностей (передняя большеберцовая, общий сгибатель пальцев кисти) выявлены слабые признаки миопатического типа поражения двигательной единицы, характеризующегося снижением длительности до 24 % от нормы, снижением амплитуды потенциалов двигательной единицы до 30 % от нормы, нарастанием фазности потенциалов до 12 %; гистограмма распределения по длительности растянута.
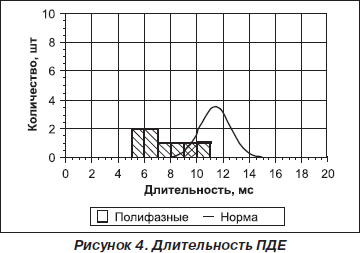
УЗДГ: снижен кровоток по ПА, больше справа. Перегруженность в бассейне ВЯВ, ПВС. Гипоперфузия по мозговым сосудам, больше слева, основной артерии.
ДС магистральных артерий головы: просвет ОСА — ВСА чистый, контуры ровные, КИМ 1.1, кровоток магистральный, симметричный. Умеренная экстравазальная компрессия ПА.
Окулист: ОИ — частичный птоз; глаза спокойны; среды прозрачны. На глазном дне: ДЗН розовые, границы четкие.
ЭКГ: синусовый регулярный ритм — 82 уд/мин. Вертикальное положение ЭОС.
Эхокардиографическое обследование: открытое овальное окно, гемодинамически незначимое. Аберрантная хорда в полости левого желудочка.
Терапевт: данных в пользу патологии внутренних органов нет.
С учетом жалоб, клинической картины, результатов дообследования проведен дифференциальный диагноз с рядом нейромышечных заболеваний.
Отсутствие положительной реакции на прозериновый тест, патологии легких, данные ЭНМГ, СКТ органов грудной клетки позволили исключить заболевания с поражением на уровне нервно-мышечного синапса (миастению гравис, миастенический синдром Ламберта — Итона).

Возраст дебюта заболевания у наблюдаемого пациента, полученные данные клинического, лабораторного и инструментального обследований, отсутствие фасцикуляций, тяжесть и скорость прогрессирования заболевания, последовательность и группы вовлекаемых мышц с развитием мышечных атрофий позволили исключить целый ряд нейрогенных заболеваний, включая болезнь двигательного нейрона, БАС, спинальную амиотрофию Кугельберга — Веландера, синдром Кенеди, невральную перонеальную амиотрофию Шарко — Мари — Туса, спинальную амиотрофию Эмери — Дрейфуса (ретракцию ахилловых сухожилий), полирадикулопатии, синдром Гийена — Барре, нейропатии при интермиттирующей порфирии, семейную гипертрофическую невропатию Дежерина — Сотта.
За время пребывания в стационаре проведен дифференциальный диагноз с воспалительными миопатиями, а также разными формами наследственных миотонических и парамиотонических синдромов и миоплегии.
От обследования в медико-генетическом центре и мышечной биопсии, к сожалению, пациент отказался.
Учитывая клиническую картину, динамику заболевания за прошедший период, данные неврологического статуса, результаты проведенных лабораторных и инструментального дообследований, пациенту установлен диагноз «прогрессирующая окулофарингеальная мышечная дистрофия с выраженными глазодвигательными нарушениями, бульбарным синдромом, выраженным амиотрофическим синдромом, миогенным тетрапарезом, выраженным нарушением функции глотания, фонации, ходьбы (G71.0)».
Назначена симптоматическая терапия: карниэль 2,0 мг 2 раза внутрь — длительно, цитофлавин, мильгамма, дибазол 5 мг 3 раза в день, нейромедин. За время пребывания в отделении, с учетом ЭНМГ-показателей, проводимой терапии, состояние пациента стабилизировалось, улучшилась фонация. При контроле через 2 месяца — состояние стабильное.
Опасность окулофарингеальной миодистрофии состоит в прогрессировании с нарастающей дисфагией и требует применения методов паллиативной неврологии. Паллиативная неврология предусматривает зондовое питание или наложение стомы. Эффективного лечения на данный момент нет. Описаны методики рассечения перстнеглоточной мышцы для улучшения глотания, но не предотвращения аспирации. Если птоз мешает зрению, используют специальные скотчевые наклейки на веки, проволочные держатели век, которые крепятся к оправе очков, либо, если нет выраженной слабости мимических мышц, прибегают к хирургическому лечению (см. http://medbiol.ru/medbiol/har/00391441.htm).
Описанный клинический случай представляет интерес не только для неврологов, но и для врачей общей практики, которые должны обращать внимание на подобные жалобы, выраженные прогрессирующие мышечные изменения, данные лабораторного и инструментального дообследования для своевременного распознавания относительно редкого и в более позднем возрасте дебютирующего заболевания — ОФМД.
Вместе с тем многолетний опыт работы в клинике показывает, что прогрессирующие мышечные заболевания у взрослых дебютируют позже и пропускаются неврологами, чаще трактуются как текущие нейроинфекции. К сожалению, рост прогрессирующих мышечных дистрофий и спинальных мышечных атрофий отмечается не только у детей, но и у взрослых. Описанный случай иллюстрирует тот факт, что подобные заболевания надо подозревать раньше. Ведь арсенал медикаментозной и немедикаментозной паллиативной неврологии достаточно большой. Хотя адекватно назначенная терапия не излечит больного, но улучшит качество его жизни и продлит саму жизнь. Кроме того, своевременная диагностика даст возможность ранней профориентации пациента.
1. Горбунова В.Н., Савельева-Васильева Е.А., Красильников В.В. Молекулярная неврология. Часть 1. Заболевания нервно-мышечной системы. — СПб.: Интермедика, 2000. — 320 с.
2. Казаков В.М. Клинико-молекулярно-генетическая классификация мышечных дистрофий (научный обзор с комментариями) // Неврол. журнал. — 2001. — № 3. — С. 47-52.
3. Евтушенко С.К., Шаймурзин М.Р., Евтушенко О.С., Евтушенко Л.Ф., Дегонская Е.В., Евтушенко И.С., Сохань Д.А.. Ранняя клинико-инструментальная диагностика и терапия быстро- и медленнопрогрессирующих мышечных дистрофий и амиотрофий // Международный неврологический журнал. — 2007. — № 4(14). — С. 47.
4. Справочник по формулированию клинического диагноза болезней нервной системы / Под ред. В.Н. Штока, О.С. Левина. — М.: ООО «Медицинское информационное агентство», 2006. — 520 с.
5. Brais B., Bouchard J.-P., Xie Y.-G., Rochefort D.L., Chretien N., Tome F.M.S., Lafreniere R.G., Rommens J.M., Uyama E., Nohira O., Blumen S., Korcyn A.D., Heutink P., Mathieu J., Duran-ceau A., Codere F., Fardeau M., Rouleau G.A. Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy // Nature Genet. — 1998. — 18. — 164-167.
6. Brais B., Xie Y.-G., Sanson M., Morgan K., Weissenbach J., Korczyn A.D., Blumen S.C., Fardeau M., Tome F.M.S., Bou-chard J.-P., Rouleau G.A. The oculopharyngeal muscular dystrophy locus maps to the region of the cardiac alpha and beta myosin heavy chain genes on chromosome 14q11.2-q13 // Hum. Molec. Genet. — 1995. — 4. — 429-434.
7. Fried K., Arlozorov A., Spria R. Autosomal recessive oculopharyngeal muscular dystrophy // J. Med. Genet. — 1975. — 12. — 416-418.
8. Goh K.J., Wong K.T., Nishino I., Minami N., Nonaka I. Oculopharyngeal muscular dystrophy with PABPN1 mutation in a Chinese Malaysian woman // Neuromusc. Disord. 2005. — 15. — 262-264.
9. Hino H., Araki K., Uyama E., Takeya M., Araki M., Yoshinobu K., Miike K., Kawazoe Y., Maeda Y., Uchino M., Yamamura K. Myopathy phenotype in transgenic mice expressing mutated PABPN1 as a model of oculopharyngeal muscular dystrophy // Hum. Molec. Genet. — 2004. — 13. — 181-190.
10. Neuromuscular disorders: gene location // Neuromusc. Disord. — 1999. — Vol. 9, № 5. — P. 1-8.
11. Robinson D.O., Wills A.J., Hammans S.R., Read S.P., Sillibourne J. Oculopharyngeal muscular dystrophy: a point mutation which mimics the effect of the PABPN1 gene triplet repeat expansion mutation // J. Med. Genet. — 2006. — 43. — e23 (Note: Electronic Article).
12. Victor M., Hayes R., Adams R.D. Oculopharyngeal muscular dystrophy. A familial disease of late life characterized by dysphagia and progressive ptosis of the eyelids // New Eng. J. Med. — 1962. — 267. — 1267-1272.