
Газета «Новости медицины и фармации» 11 (421) 2012
Вернуться к номеру
Ураження нирок при системних хворобах
Авторы: А.Н. Корж, д.м.н., професор, завідувач кафедри загальної практики — сімейної медицини ХМАПО, член Всесвітньої організації сімейних лікарів WONCA; Д.Д. Іванов, д.м.н., професор, заслужений лікар України, завідувач кафедри нефрології і нирково-замісної терапії НМАПО імені П.Л. Шупика
Версия для печати
Ураження нирок (нефропатії, нефрити або хронічні хвороби нирок), що виникають при ревматичних хворобах, включають цілу низку нозологічних форм, які до сьогодні не мають визнаної класифікації. Згідно з відомою монографією (Н.А. Колесник с соавт., 2004) та світовими джерелами, виділяють ураження нирок, обумовлені системними васкулітами, системними захворюваннями сполучної тканини, ревматичними артритами, ревматизмом і мієломною хворобою (рис. 1).
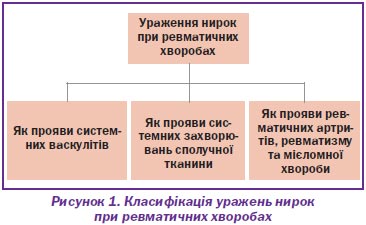
У дебюті або протягом перебігу ревматичного захворювання виникають ураження нирок. Цей стан, що кваліфікується як прояв основного захворювання та як хронічна хвороба нирок, потребує консультації, спостереження, а при прогресуванні — лікування в нефролога. Діагностика таких ХХН є не завжди доступною, перебіг — хвилеподібним, лікування — тривалим та не дуже успішним. Базовою терапією є ІАПФ, що призначаються на все життя, сартани та препарати, які покращують реологічні властивості крові. Основою лікування є глюкокортикоїди й цитостатики, за наявності термінальної ХНН — діаліз. Через ураження судин при васкуліті перитонеальний діаліз є методом вибору в лікуванні таких пацієнтів. Трансплантація нирок у більшості випадків ефективна. Спостереження і ведення таких хворих проходять на мультидисциплінарній основі.
Ураження нирок, обумовлені системними васкулітами
Васкуліти розділяють (Chapel Hill, 1998) на великосудинні (гігантоклітинний артеріїт — хвороба Хортона; неспецифічний аортоартеріїт Такаяші), середньосудинні (вузликовий поліартеріїт — поліартеріїт нодоза, хвороба Кавасакі) та дрібносудинні. Останні, у свою чергу, поділяються на васкуліти з імунними комплексами (СЧВ, геморагічний васкуліт — пурпура Шенлейна — Геноха, кріоглобулінемія, а також хвороба Бехчета та ураження нирок при ревматоїдному артриті) та автоімунний ANCA-позитивний васкуліт (гранулематоз Вегенера, мікроскопічний поліангіїт та синдром Чарга — Стросса). Окремо виділяють васкуліти, що асоціюються з органоспецифічними антитілами: синдром Гудпасчера й середньосудинний васкуліт (хвороба Кавасакі).
У 2002 році Європейською групою з вивчення васкулітів (EUVAS) запропонована така класифікація для автоімунних системних васкулітів:
- Локалізовані безсимптомні васкуліти, що не впливають на функцію життєво важливих органів, креатинін крові менше ніж 120 мкмоль/л, ANCA-позитивний/негативний.
- Ранні системні васкуліти, що не порушують функцію життєво важливих органів, креатинін крові менше ніж 120 мкмоль/л, ANCA-позитивний/негативний.
- Генералізовані системні васкуліти, що супроводжуються дисфункцією життєво важливих органів, креатинін крові менше ніж 500 мкмоль/л, ANCA-позитивний.
- Тяжкий нирковий васкуліт із загальними симптомами, креатинін крові понад 500 мкмоль/л, ANCA-позитивний.
- Рефрактерний інкурабельний васкуліт.
У нефрологічній практиці найбільш небезпечними є васкуліти дрібних судин, що нерідко супроводжуються розвитком півмісяців і, відповідно, необоротною втратою функції нирок і формуванням термінальної хронічної ниркової недостатності. Причини, що призводять до склерозування нирок із півмісяцями, наведені на рис. 2.
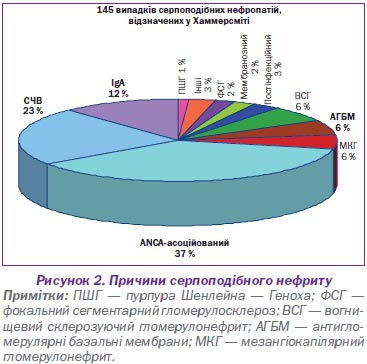
Як свідчать дані рис. 2, найбільша частота несприятливих для життя ренальних васкулітів припадає на дрібносудинні ураження, а саме автоімунні ANCA-позитивні хвороби — мікроскопічний поліангіїт, гранулематоз Вегенера та синдром Чарга — Стросса і швидкопрогресуючий гломерулонефрит. Це цілком зрозуміло, тому що нирки мають найбільшу капілярну систему організму. На підставі субтипів ANCA проводять диференціальну діагностику між наявністю антитіл до протеїнази-3 (PR3) — pANCA і мієлопероксидази (МРО) — cANCA (рис. 3).
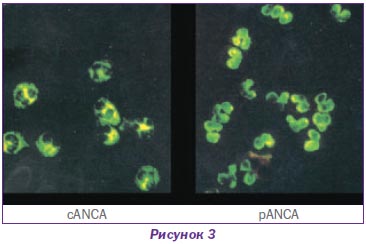
Для пацієнтів із гранулематозом Вегенера характерні анти-PR3 антитіла, для мікроскопічного поліангіїту — анти-МРО. Анти-PR3 захворювання характеризуються високою активністю процесу зі швидким розвитком ниркової недостатності, в основі якого є некроз, епітеліальні півмісяці. Але за наявності адекватної терапії можливе відновлення функції, хоча й існує більш висока можливість рецидиву. Мікроскопічний поліангіїт з анти-МРО характеризується більш спокійним перебігом із невисокою активністю захворювання, прогресуючим перебігом із втратою функції, низьким ризиком рецидивів та подальшим морфологічним субстратом: півмісяці, гломерулосклероз, інтерстиціальний фіброз.
Гранулематоз Вегенера
Код за МКХ-10 — М 31.3+N08.5
Морфологічні зміни:
- Фокально-сегментарний некротизуючий ГН.
- Дифузний проліферативний ГН.
- Мембранопроліферативний ГН.
- ГН із півмісяцями.
- Гранулематозний ГН.
Приклад діагнозу:гранулематоз Вегенера, ХХН 3-ї стадії: мембранопроліферативний ГН, артеріальна гіпертензія 1-го ступеня, анемія 1-го ступеня.
Патогномонічною ознакою є виявлення ANCA з наявністю антитіл до протеїнази-3.
При гранулематозі Вегенера спостерігається ураження судин верхніх і нижніх дихальних шляхів, що супроводжується розвитком фарингіту, синуситу, бронхіту, пневмоніту, та нирок — нефропатії із клінічною картиною гломерулонефриту. Серед менш частих проявів гранулематозу Вегенера слід відзначити кожні висипки, артрити, коронарити, кишкові кровотечі. Вважають, що для встановлення діагнозу необхідні щонайменше дві ознаки з таких:
- виразкові ураження слизової оболонки носа або ротової порожнини;
- вузликові утворення або фіксовані інфільтрати легенів на рентгенограмі;
- еритроцитурія;
- гранульоми в біоптаті.
Мікроскопічний поліангіїт
Код за МКХ-10 — М 31.8+N08.5
Морфологічні зміни:
- Фокальний сегментарний некротизуючий ГН.
- Дифузний ГН із півмісяцями.
Приклад діагнозу: Мікроскопічний поліангіїт, ХХН 4-ї стадії: фокальний сегментарний некротизуючий ГН, артеріальна гіпертензія 3-го ступеня, анемія 2-го ступеня.
Патогномонічною ознакою є виявлення ANCA з наявністю антитіл до мієлопероксидази.
Мікроскопічний поліангіїт є некротизуючим васкулітом, що характеризується наявністю легеневих геморагій, прогресуючим перебігом ГН із загостреннями і зростанням порушення функції нирок та рідко полінейропатією.
Терапія гранулематозу Вегенера та мікроскопічного поліангіїту повинна проводитись у нефрологічному відділенні й передбачає: 1) застосування невеликих доз преднізолону з тривалим призначенням цитостатиків; 2) призначення пульс-терапії метипредом і пульс-терапії цитостатиком; 3) використання мофетилу мікофенолату та інфліксимабу (антитіл до тумор-некротизуючого фактора — Remicade).
До автоімунних ANCA-позитивних дрібносудинних васкулітів відноситься також синдром Чарга — Стросса. Класифікаційними критеріями синдрому є:
- астма та обтяжений алергологічний анамнез;
- еозинофілія (понад 10 %) із позасудинними скупченнями еозинофілів (за результатами біопсії);
- моно-/полінейропатія;
- легеневі інфільтрати;
- синусити.
До частих проявів синдрому належить ураження ЛОР-органів (поліпоз, вазомоторний риніт), кишечника (неспецифічний виразковий коліт), серця (ендо- й перикардит, гіпертензія), шкіри (пурпура, еритема), суглобів (артрити), нервової системи (енцефалопатія) та нирок. Ураження нирок спостерігається в 6–10 % пацієнтів, характеризується мезангіопроліферативним ГН із поступовим розвитком ХНН. До ранніх ознак ураження нирок відноситься нормоцитарна нормохромна анемія, зниження питомої ваги сечі та розвиток діастолічної гіпертензії. Патогномонічною ознакою є виявлення р-ANCA.
Лікування синдрому Чарга — Стросса передбачає призначення глюкокортикоїдів і цитостатиків.
До дрібносудинних васкулітів з імунними комплексами відносяться СЧВ, геморагічний васкуліт — пурпура Шенлейна — Геноха, кріоглобулінемія та хвороба Бехчета.
Системний червоний вовчак (СЧВ)
Вовчаковий нефрит (ВН), або люпус-нефрит,— ураження нирок при СЧВ. Клініцист повинен добре знати це серйозне захворювання, тому що при правильному лікуванні прогноз у хворих на ВН може бути значно поліпшеним.
СЧВ — найчастіше системне захворювання, що уражає нирки. Поширеність СЧВ у європейській популяції — 30–40 на 100 000. Хворіють переважно жінки віком 20–40 років; провокуючими факторами можуть бути інсоляція, медикаментозна непереносимість, стрес, вагітність. У чоловіків СЧВ спостерігається значно рідше (співвідношення жінок і чоловіків становить 8–9 : 1).
У розвитку захворювання відіграють роль як ендогенні (генетичні, статеві гормони), так і екзогенні (віруси, УФ-опромінення) фактори. Є дані про істотну роль спадковості. СЧВ зустрічається в різних етнічних групах, але особливо часто в афро-карибській популяції (до 200 на 100 000). У США поширеність СЧВ і смертність від нього серед чорношкірих жінок у 10 разів вищі, ніж серед представниць білої раси. Частота захворювання родичів хворих (4–50 на 1000) у кілька разів вища, ніж у популяції. Неодноразово описувалися сімейні випадки хвороби зі схожою клінічною картиною, у тому числі в близнюків.
Серед впливів зовнішнього середовища в розвитку СЧВ передбачається (хоча і не доведена) роль вірусів. У тканині нирок хворих на ВН виявлені структури, що нагадують скупчення параміксовірусів.
У даний час увагу дослідників привертає підвищення титрів до вірусів, що містять РНК, у сироватці хворих та експериментальних тварин, а також ретровіруси. Вірусну етіологію СЧВ опосередковано підтверджують і дослідження, що показали значно більшу частоту високих титрів антитіл до вірусу кору (РНК-умісний параміксовірус), а також корового геному в лімфоцитах хворих із високими титрами антитіл. Описано розвиток ВН після парвовірусної інфекції.
Роль вірусів при СЧВ останнім часом обговорюється й у зв’язку з виявленням подібності імунних порушень і деяких клінічних симптомів при СЧВ і СНІДі: лімфоцитопенія, зниження рівня Т-хелперів, підвищення рівня ЦІК. ВІЛ ушкоджує клітини, що експресують вірусний рецепторний білок CD4, який є маркером Т-лімфоцитів CD4, що відіграють центральну роль у розвитку імунної відповіді. При ВІЛ у сироватці виявляються антинуклеарні фактори, антитіла до фосфоліпідів, підвищений уміст g-глобулінів, спостерігається вовчакоподібний синдром (гарячка, нездужання, схуднення, протеїнурія, гематурія, ниркова недостатність, тромбоцитопенія, лейкопенія, лімфопенія).
Патогенез
СЧВ — генералізоване автоімунне захворювання, при якому виявляються численні антитіла до власних антигенів, у першу чергу до ядерних антигенів — нуклеїнових кислот і білків, що мають відношення до внутрішньоклітинної передачі інформації. Найбільш характерні для СЧВ антитіла до нативної ДНК і нуклеосом (комплексу «ДНК — гістон»). Серед безлічі інших антитіл виділяють антитіла до кардіоліпіну, виявлення яких часто поєднується з антифосфоліпідним синдромом (що спостерігається і поза СЧВ), у тому числі з нирковими артеріальними, венозними й капілярними (клубочковими) тромбозами, ендокардитом і цереброваскулітом.
Причина утворення антитіл — зниження толерантності до власних антигенів (неефективна толерантність, порушення розпізнавання антигенів), дефект Т-системи (зниження активності Т-супресорів) і В-системи (поліклональна активація). Патогенез нефриту пов’язують із відкладенням у нирках імунних комплексів, що містять ядерні антигени й антитіла до них. ВН розглядається як класична модель імунокомплексного нефриту.
Довгий час основне патогенетичне значення надавалося системі «ДНК — антитіла до ДНК». Однак у даний час як автоантиген, що починає імунну відповідь при СЧВ, розглядають нуклеосоми. Центральна роль нуклеосом підкреслюється їх більш раннім порівняно з іншими антиядерними антитілами, у тому числі антитілами до ДНК, утворенням.
Вовчакові автоантитіла гетерогенні щодо їх антигензв’язуючої специфічності й ушкоджуючого потенціалу (пов’язаних між собою). Розвиток нефриту у хворого на СЧВ залежить від наявності автоантитіл, що мають нефритогенні властивості. Різні антитіла значно відрізняються за здатністю локалізуватися в нирках і/або викликати функціональні зміни. Ig, ізольований із сироватки хворих на ВН, при перфузії через ізольовану нирку щура може зв’язуватися з клубочками на відміну від Ig, отриманого від хворих на СЧВ без ураження нирок.
ВН — найбільш тяжкий вісцерит при СЧВ, часто визначає прогноз хвороби, зустрічається в 50–70 % хворих, звичайно розвивається на 2–3-му році захворювання.
Морфологія ВН відрізняється значним поліморфізмом. Відзначаються проліферація клітин клубочків, розширення й інтерпозиція мезангіуму, мембранозні зміни, ураження канальців й інтерстицію, склероз судинних петель; характерною є саме розмаїтість змін як в одному, так і в різних клубочках. Специфічними (хоча й не патогномонічними) для ВН морфологічними ознаками вважаються фибриноїдний некроз капілярних петель, ядерна патологія — каріорексис і каріопікноз, різке вогнищеве стовщення базальних мембран капілярів клубочків у вигляді «дротових петель». Важливим елементом ушкодження є внутрішньосудинний тромбоз (фібринові й гіалінові тромби у просвіті капілярів), що, можливо, поєднуються з наявністю антикардіоліпінових антитіл, хоча деякі автори вважають, що гіалінові тромби — відкладення імунних комплексів, що містять кріоглобуліни.
Розрізняють вогнищевий і дифузний ВН (при наявності специфічних ознак), мембранозний гломерулонефрит (Г), а також мезангіопроліферативний, мезангіокапілярний і фібропластичний ГН.
За класифікацією ВООЗ виділяють такі морфологічні типи ВН: нормальні клубочки (клас I); тільки мезангіальні зміни — розширення або гіперклітинність (клас II); осередковий ГН з активними некротизуючими і/або склеротичними змінами, з ураженням менше ніж 50 % клубочків (клас III); дифузний ГН із такими ж змінами, але із залученням більше ніж 50 % клубочків (клас IV); мембранозний ГН (клас V) і склерозуючий ГН (клас VI).
Системний червоний вовчак з ураженням нирок класифікується за МКХ-10: М32.1+N08.5
Морфологічна характеристика (класи) люпус-нефриту (ЛН):
І. Мінімальний мезангіальний ЛН.
ІІ. Мезангіальний проліферативний ЛН.
ІІІ. Фокальний ЛН (ушкоджені менше ніж 50 % всіх клубочків).
ІІІ (А). Фокальний проліферативний ЛН.
ІІІ (А/С). Фокальний проліферативний та склерозуючий ЛН.
ІІІ (С). Фокальний склерозуючий ЛН.
ІV. Дифузний сегментарний (IV-S) або глобальний (IV-G) ЛН (ушкоджені понад 50 % всіх клубочків).
IV-S(А). Дифузний сегментарний проліферативний ЛН.
IV-G(А). Дифузний глобальний проліферативний ЛН.
IV-S(А/С). Дифузний сегментарний проліферативний та склерозуючий ЛН.
IV-G(А/С). Дифузний глобальний проліферативний та склерозуючий ЛН.
IV-S(С). Дифузний сегментарний склерозуючий ЛН.
IV-G(С). Дифузний глобальний склерозуючий ЛН.
V. Мембранозний ЛН.
VІ. Прогресуючий склерозуючий ЛН (понад 90 % усіх клубочків глобально склерозовані):
А) вказується частка клубочків з активними та склеротичними змінами;
В) вказується частка клубочків із наявністю фібриноїдного некрозу та/або клітинних півмісяців;
С) клас V може бути поєднаний з класами ІІІ та/або ІV.
Приклади діагнозу: СЧВ, ХХН 5-ї стадії, люпус-нефрит VІ класу (прогресуючий склерозуючий ЛН із вираженими тубулярною атрофією, інтерстиціальним фіброзом та артеріосклерозом), артеріальна гіпертензія 3-го ст., анемія 2-ї ст.
СЧВ, ХХН 3-ї стадії, люпус-нефрит ІV-G(А/С) класу (дифузний глобальний проліферативний та склерозуючий ЛН із наявністю клітинних півмісяців у 50 % клубочків, помірною тубулярною атрофією, вогнищевим інтерстиціальним фіброзом та розвитком інтимального артеріосклерозу), нефротичний синдром, артеріальна гіпертензія 2-го ст., анемія 1-ї ст.
Клінічна картина ВН так само різноманітна, як і морфологічна, — від мінімальної персистуючої протеїнурії, що не впливає на прогноз, до найтяжчого швидкопрогресуючого ВН (ШПВН), із набряками, анасаркою, нирковою недостатністю. Немає яких-небудь ознак, властивих тільки ВН, хоча в деяких випадках особливості клінічного перебігу змушують запідозрити саме вовчакову природу ураження нирок. Характерні помірна протеїнурія в поєднанні з еритроцитурією, у той же час ізольована протеїнурія, як і ізольована гематурія (тим більше макрогематурія), зустрічається рідко. Нефротичний синдром звичайно сполучається з гематурією й АГ, протеїнурія рідко буває вираженою, гіпоальбумінемія, гіпер-a2-глобулінемія й гіперхолестеринемія помірні. АГ частіше розвивається при вираженому сечовому синдромі, здебільшого (особливо у хворих без нефротичного синдрому) поєднується з антикардіоліпіновими антитілами. Досить часто має місце ШПВН, що супроводжується внутрішньосудинним згортанням.
Клінічна картина деякою мірою відповідає морфологічній: швидке прогресування найбільш характерне для дифузного ВН, тоді як при мезангіальних і мембранозних змінах функція нирок звичайно довгостроково збережена.
Залежно від тяжкості клінічної картини, перебігу і прогнозу виділяють такі клінічні варіанти ВН:
- швидкопрогресуючий ВН;
- активний ВН із нефротичним синдромом;
- активний ВН із вираженим сечовим синдромом;
- неактивний ВН;
- латентний ВН.
Для активного ураження нирок характерні: наростання протеїнурії, нефротичний синдром, АГ, еритроцитурія, швидке погіршення функції нирок, високий титр антитіл до нативної ДНК, а також тромбоцитопенія, анемія та зниження рівня комплементу. Шість останніх показників найбільш точно прогнозують поганий результат, особливо якщо вони виявляються на ранніх етапах хвороби.
Діагностика
Діагноз ВН у більшості випадків установлюється за наявності характерних клінічних і лабораторних ознак СЧВ: найчастіше це артралгії й артрити (недеформуючі) дрібних суглобів, особливо кистей, і шкірні висипання, серед яких діагностичне значення має вовчаковий «метелик» — чітко окреслена еритема на щоках і переніссі. Характерні плеврит, пневмоніт, перикардит, ендокардит (частіше аортальних клапанів), ураження нирок і ЦНС, утрата маси тіла, гарячка.
Із лабораторних ознак для СЧВ характерні лейкопенія, анемія, тромбоцитопенія, підвищення ШОЕ, зниження титру комплементу (у хворих ВН), гіпер-g-глобулінемія. Діагностично значиме виявлення антитіл до ядерних компонентів — до нативної ДНК, гістонів і до структур рибонуклеопротеїну. У хворих із ВН антитіла до нативної ДНК виявляються частіше, ніж у хворих без нефриту. Sm-антитіла високоспецифічні для ВН, але виявляються тільки в 15–20 % хворих.
При розгорнутій картині СЧВ встановлення діагнозу ВН не викликає труднощів, за винятком випадків, коли захворювання починається в молодих жінок гарячково-шкірно-суглобовим синдромом із підвищеною ШОЕ, лейкопенією, перебігає з періодичними загостреннями, на висоті одного з яких приєднується плеврит (або перикардит); одночасно або через 1–2 роки з’являється протеїнурія. Вірогідний діагноз повинен бути підтверджений наявністю антитіл до ДНК або антинуклеарного фактора (ANA) та Sm-антитіл. За відсутності суглобного синдрому й імунологічних доказів діагноз потрібно встановлювати з великою обережністю, особливо в чоловіків. Біопсія нирки допомагає уточнити морфологічний варіант ВН, особливо при персистуючій протеїнурії або персистуючому нефротичному синдромі (виявлення мембранозного ГН дозволяє проводити менш активну терапію), а при повільному зниженні функції нирок вирішити, чи йдеться про активний процес або про поступове склерозування нирок.
Диференціальну діагностику проводять із підгострим бактеріальним ендокардитом, ревматоїдним артритом, множинною мієломою, хронічним активним гепатитом, первинним і спадковим амілоїдозом, пурпурою Шенлейна — Геноха.
Прогноз
Прогноз у таких хворих, що залежить як від тяжкості ураження нирок, так і від своєчасного призначення активної терапії, значно покращився в останні десятиліття. 30–40 років тому лише одиничні хворі з тяжким ВН жили більше ніж 1–2 роки, при менш тяжких формах половина хворих помирали в найближчі 5 років. У даний час 70–80 % хворих із тяжким ВН живуть довше ніж 5 років.
Лікування
Лікування ВН проводиться постійно, довгостроково і включає два етапи: 1) терапію гострого дебюту/загострення, що часто перебігає з ураженням багатьох органів (звичайно незабаром після початку хвороби); мета терапії — зменшити активність процесу, тобто індукувати ремісію, зупинити прогресування ВН, домогтися ремісії або хоча б поліпшення (період індукції ремісії); 2) тривалу підтримуючу терапію. В обидва періоди основу лікування становлять кортикостероїди й цитостатики.
1-й етап. Терапія індукції. Лікування активної фази повинне починатися якомога раніше. Іноді затримка з лікуванням навіть на 1–2 тижн. (а при швидкому прогресуванні — на 4–5 днів) уже не дозволяє домогтися ремісії.
При невисокій активності нефриту можна обмежитися преднізолоном у дозі 60 мг/добу протягом 2 міс. із подальшим дуже повільним зниженням дози до підтримуючої. Серед препаратів 3-го морфологічного класу застосовують пульс-терапію метилпреднізолоном 0,75–1 г/добу щоденно (через добу) № 3–12 у поєднанні з пульс-терапією ЦФ у дозі 10 мг/кг 1 раз на 2 тижні або 20 мг/кг один раз на місяць (сумарна доза 3 г). Альтернативою ЦФ виступає мофетилу мікофенолат у дозі 2–3 г/добу. Така тактика базується на результатах десятирічного спостереження за хворими в дослідженні EURO-LUPUS (Ann. Rheum. Dis. — 2010 Jan. — 69(1). — 61-4). Зниження ШКФ менше 30 мл/хв потребує проведення імунофорезу.
2-й етап. Терапія підтримання. Після значного клінічного поліпшення знижувати дозу метилпреднізолону слід на 2 мг кожні 12 тижнів, призначаючи препарат через добу. Серед цитостатиків пере-вага надається мофетилу мікофенолату (доза постійна протягом всього лікування) або азатіоприну (100–200 мг/добу).
За наявності високої активності люпус-нефриту при резистентності до призначеної терапії застосовують ритуксимаб по 1 г двічі протягом 2 тижнів один раз на півроку.
Підтримуюча терапія проводиться роками (у всякому разі не менше ніж 2 роки, але звичайно значно довше), при цьому особливого значення набуває профілактика побічних ефектів. Основою підтримуючої терапії є преднізолон (10–20 мг) і цитостатики. Терапія ГК і цитостатиками завжди поєднується з використанням інгібіторів АПФ.
У 10–15 % хворих розвивається термінальна ниркова недостатність. Гемодіаліз і трансплантація нирок дозволяють значно збільшити тривалість життя таких хворих.
У перші роки застосування гемодіалізу при ВН смертність була значно вищою, ніж при первинному ГН. Однак в останнє десятиліття результати цього лікування значно покращилися і 5-річна виживаність при ВН після початку гемодіалізу становила в Австралії, США й у Данії 81–89 %, тобто стала такою ж, як при інших захворюваннях нирок. У той же час частина пацієнтів, які знаходяться на гемодіалізі, мають потребу в імуносупресивній терапії.
Трансплантація нирок показана при розгорнутій клінічній картині уремії, активність СЧВ до цього часу звичайно зменшується, тому страх перед загостренням СЧВ із рецидивом ВН у трансплантаті не обґрунтований.
Таким чином, на рубежі століть ВН хоча і є тяжким захворюванням, але піддається тривалій активній терапії. При розвитку прогресуючого нефриту у молодих жінок навіть за відсутності характерних екстраренальних симптомів варто мати на увазі можливість СЧВ, ретельно збирати анамнез (ефемерні артралгії, фотосенсибілізація й ін.) та аналізувати лабораторні дані (цитопенія, зниження рівня комплементу). При підтвердженні діагнозу (як і при розвитку ураження нирок в осіб із раніше установленим СЧВ), можливо, варто раніше починати максимально активне лікування. Замісна ниркова терапія (діаліз, трансплантація нирок) у хворих на ВН із термінальною нирковою недостатністю дає такі ж результати, як і у хворих із хронічним нефритом.
Геморагічний васкуліт
Геморагічний васкуліт з ураженням нирок (Шенлейна — Геноха пурпура)
Код за МКХ-10 — І77.8+N08.5
Класи морфологічних змін
Приклад діагнозу:геморагічний васкуліт, ХХН 3-ї стадії: псевдомембранозно-проліферативний ГН (клас VІ), артеріальна гіпертензія 2-го ступеня, анемія 2-го ступеня.
Геморагічний васкуліт є досить поширеною хворобою порівняно з іншими васкулітами. Класифікаційно виділяють шкірну та змішану форми. Остання супроводжується ураженням кишечника, нирок та суглобів.
Клінічними ознаками геморагічного васкуліту, що розвивається гостро, є розвиток захворювання в дитячому або молодому віці, пурпура, що пальпується, та петехії, артралгії та кишковий біль (абдомінальний синдром), гематуричний варіант гломерулонефриту та гіпертензія. Типовою є стадійність захворювання, що починається із шкірних проявів та обтяжується ураженням інших систем. Наявність васкуліту головного мозку та легеневого васкуліту не є типовою для захворювання. Ураження нирок визначає прогноз геморагічного васкуліту.
За наявності гематуричної форми гломерулонефриту прогноз є більш сприятливим, ніж при супутній протеїнурії. Будь-який ступінь протеїнурії визначає необхідність призначення ІАПФ/сартанів або їх комбінації. Високий рівень протеїнурії (нефротичний синдром) або раннє зниження питомої ваги сечі свідчать про необхідність активного (агресивного) ведення таких хворих. Глюкокортикоїдна терапія не є високоефективною, тому цитостатики визначають безпосередній перебіг геморагічно-го васкуліту, а ІАПФ/сартани — довготривалий прогноз.
Кріоглобулінемія
Кріоглобулінемія — це феномен преципітації патологічних імуноглобулінів у гель при температурі 4 °С, що може розглядатися як синдром або самостійна нозологічна форма. При цьому розвивається імунокомплексний васкуліт, що супроводжується ураженням переважно шкіри (болісна пурпура на гомілках та стопах), артралгіями, м’язовою слабкістю та нейропатією. Відмінною характеристикою від інших васкулітів є наявність гепатитів С і В, а також частий розвиток легеневого васкуліту (кровохаркання) та синдрому Рейно. У більшості випадків ураження нирок виникає протягом перших 4 років після дебюту захворювання та проявляється мезангіокапілярним гломерулонефритом із наявністю PAS-позитивних внутрішньокапілярних тромбів. Лікування є посиндромним із застосуванням глюкокортикоїдів і цитостатиків за наявності високої активності процесу. Наявність активності гепатиту потребує призначення пегільованих інтерферонів.
Хвороба Бехчета
Хвороба Бехчета являє собою васкуліт переважно дрібних та середніх судин із розвитком гіпертензії в кожного третього пацієнта. Клінічними проявами хвороби є виразковий безболісний стоматит, виразкове ураження слизової геніталій (що супроводжується болем), вузликова еритема та папуло-пустульозна висипка, ураження слизової очей, артрити та артралгії, ураження кишкового тракту (коліт), центральної нервової системи. Ураження нирок виникає протягом перших 10–12 років від дебюту захворювання і проявляється гломерулопатією з можливим розвитком амілоїдозу та рідким прогресуванням до термінальної ХНН. У лікуванні нирок використовують глюкокортикоїди і цитостатики, за наявності амілоїдозу — колхіцин.
До середньосудинних васкулітів належать вузликовий поліартеріїт — поліартеріїт нодозний та хвороба Кавасакі.
Вузликовий періартеріїт (ВП)
Як один із різновидів системного алергійного васкуліту, він характеризується зміною судин, переважно дрібних і середніх артерій, із ураженням ряду органів і систем.
Клінічна симптоматика поліморфна, звичайно провідне місце займає патологія нирок, що зустрічається в 70–80 % випадків і в основному визначає прогноз хвороби. Хворіють переважно чоловіки віком 30–50 років.
У 30–50 % хворих на ВП у сироватці крові виявляють австралійський антиген (вірус сироваткового гепатиту) й імунологічні порушення, пов’язані з персистуванням цього антигену.
При ВП уражаються внутрішньоорганні судини нирок середнього калібру (дугові, міжчасткові, прямі артерії, що приносять і виносять клубочкові артеріоли, капіляри і вени), а іноді й основний стовбур ниркової артерії. Зміни в них мають характер продуктивного, рідше продуктивно-деструктивного панваскуліту, що завершується склерозом, стовщенням стінок (особливо інтими), утворенням аневризм, стенозуванням і тромбозом просвіту судин. Інфаркти нирок майже патогномонічні для цього захворювання. При великих інфарктах можливе зморщення нирки. Нерідко ниркові ураження виявляються минущим сечовим синдромом або дифузним гломерулонефритом. Зміни в ниркових клубочках мають мембранозний, проліферативно-мембранозний, фібропластичний або екстракапілярний характер.
Хвороба починається поступово і, як правило, із загальних симптомів: гарячки неправильного типу, схуднення, м’язово-суглобових болів. Потім відзначається вісцеральна симптоматика з п’ятьма провідними синдромами: нирковим з артеріальною гіпертензією; абдомінальним; коронарним із розвитком стенокардії або інфаркту міокарда, нерідко безболісним; легеневим, що виявляється своєрідним пневмонітом або бронхіальною астмою; поліневритичним. Із лабораторних даних важливе значення мають лейкоцитоз, підвищення ШОЕ, іноді еозинофілія.
Відповідно до Американської колегії ревматологів (1990) виділяють такі класифікаційні критерії ВП:
- зменшення маси тіла більше ніж на 4 кг від початку захворювання, не пов’язане зі зміною дієти;
- сітчасте ліведо;
- біль або болісність при пальпації яєчок;
- міалгії, слабкість або болісність у м’язах нижніх кінцівок;
- мононеврит або полінейропатія (асиметрична);
- діастолічний тиск більше ніж 90 мм рт.ст.;
- підвищення рівня креатиніну/сечовини крові;
- інфікування вірусом гепатиту В;
- аневризми або оклюзії вісцеральних артерій, не пов’язані з атеросклерозом, фібромускулярною дисплазією й іншими незапальними захворюваннями за даними артеріографічних змін;
- нейтрофіли у стінці дрібних і середніх артерій за результатами біопсії.
Розрізняють такі клінічні варіанти ниркових уражень при ВП.
Латентні форми ураження нирок при доброякісному перебігу захворювання характеризуються помірною протеїнурією з нормальним сечовим осадом, невеликими функціональними порушеннями у вигляді зменшення клубочкової фільтрації й відхиленнями при радіонуклідній реносцинтиграфії.
При ізольованому сечовому синдромі відзначаються ізольована протеїнурія (до 1 г на добу) і мікрогематурія. Це ураження нирок не прогресує, його перебіг сприятливий.
Розглянутий синдром може свідчити і про початкову стадію тяжкого ураження нирок із приєднанням артеріальної гіпертензії й ниркової недостатності.
Синдром артеріальної гіпертензії зустрічається в 45–80 % хворих. Він може розвиватися прогресивно або стабільно. У 10 % хворих цей синдром передує ураженню нирок. При наявності ниркової патології він, як правило, набуває злоякісного характеру й уже в перші роки хвороби призводить до хронічної ниркової недостатності. Відомі випадки, коли у хворих при поєднанні ниркової патології з тяжким ураженням легень розглянутий синдром був відсутнім.
Нефротичний синдром при ВП зустрічається надзвичайно рідко. Характеризується високою протеїнурією (до 15–30 г на добу), гіпопротеїнемією, незначною гіперхолестеринемією, набряками зі злоякісною артеріальною гіпертензією.
При ВП спостерігаються й рідкісні форми ураження нирок. Серед них можна виділити три форми. Перша обумовлена судинними катастрофами (розриви аневризми, некроз кори нирок та ін.). Перебігає блискавично, нерідко з розвитком гострої ниркової недостатності. Друга форма пов’язана з утворенням навколониркової гематоми в результаті розриву аневризми. Характеризується раптовою гематурією й сильними болями в попереку, іноді перебігає під маскою різної патології сечової системи. Діагностувати дану форму дозволяє екскреторна урографія. Третя форма — злоякісна тубулопатія — виникає внаслідок некрозу проксимальних відділів канальців. Характеризується поліурією (до 10 л на добу), порушенням балансу електролітів, алкалозом.
У діагностиці широко застосовується пункційна біопсія внутрішніх органів (печінка, легені, нирки) або підшкірних вузликів. Необхідно зазначити, що біопсія нирок іноді викликає небажані ускладнення, тому даний метод варто використовувати лише в крайніх випадках.
Оптимальні результати можна одержати за допомогою артеріографії. Істотна вада цього методу полягає, на жаль, у тому, що він виявляє головним чином пізні зміни, такі, що далеко зайшли. Гарний ефект при функціональному дослідженні нирок дають радіоренографія (застосовується і для тяжкохворих) і кліренс-тести. Складними для діагностики є випадки, коли ураження нирок передує іншим системним проявам ВП, при стертих формах хвороби, що перебігають з ізольованою протеїнурією або гематурією.
На сьогодні для лікування ВП використовуються кортикостероїди, імунодепресивні препарати, 4-амінохіноліни й адекватна симптоматична терапія, направлена в основному на лікування гіпертензії. Препаратами вибору у лікуванні гіпертензії є ІАПФ, сартани, кальцієві блокатори (фелодипін, лерканідипін, дилтіазем), фізіотенз.
Застосування кортикостероїдів обумовлене тим, що вони пригнічують лихоманку, суглобово-м’язовий синдром, у більшості хворих поліпшують перебіг органних васкулітів. Глюкокортикоїдні гормони призначають у середніх дозах (60 мг на добу). У міру поліпшення стану хворих дозу преднізолону поступово знижують. Підтримуюча терапія глюкокортикоїдами продовжується багато місяців і навіть роки. У той же час варто враховувати, що вони впливають на ураження нирок.
Вибір методу лікування нерідко утруднює високий артеріальний тиск. Тому у хворих зі стійкою артеріальною гіпертензією, тобто з нирково-вісцеральним або нирковим варіантом ВП, доцільно поєднувати стероїдну терапію й цитостатики (азатіоприн, імуран, циклофосфамід). Показаннями до застосування імунодепресантів є резистентність або погіршення стану хворого при лікуванні преднізолоном і генералізовані форми захворювання з ураженням нирок. Застосування кортикостероїдів й імунодепресантів можливе й у вигляді пульс-терапії. У дебюті захворювання ефективні також методи екстракорпорального очищення крові — гемосорбція й плазмаферез.
При тромбоангіотичному варіанті ВП рекомендується комплексна терапія із застосуванням кортикостероїдів, антикоагулянтів і симптоматичних засобів (тиклопідин, глікозаміноглікани, ензими й ін.). Варто уникати введення білкових препаратів, плазми крові у зв’язку з можливими алергійними реакціями й загостренням захворювання.
Профілактика ураження нирок при ревматичних захворюваннях і ВП — це рання діагностика нозологічних форм із детальним і глибоким клініко-лабораторним обстеженням і регулярним диспансерним спостереженням хворих.
Диспансеризацію цього контингенту хворих здійснює лікар-ревматолог, при його відсутності — сімейний лікар або дільничний терапевт, при необхідності консультують нефролог та інші фахівці. Лікар оглядає хворих 2–4 рази на рік (при цьому проводиться клінічний аналіз крові і загальний аналіз сечі). За показаннями призначають необхідні лабораторні (у тому числі визначення рівнів сечовини і креатиніну крові), рентгенологічні і функціонально-діагностичні дослідження. Усі диспансеризовані хворі повинні одержувати протирецидивне амбулаторне, а за необхідності і стаціонарне лікування. Не менш важливим є і контроль за працевлаштуванням хворих.
Хвороба Кавасакі
Васкуліт судин середнього калібру характерний для мешканців Японії та зустрічається переважно в новонароджених та дітей молодшого віку. Захворювання починається гостро й характеризується стійкою до антибіотикотерапії лихоманкою, ураженням слизових оболонок кон’юнктиви (двобічний кон’юнктивіт), ротової порожнини (малиновий язик), губ, шкіри (еритема долонь, підошов, десквамація кінчиків пальців, поліморфна екзантема тулуба) та супутнім лімфаденітом і нерідко діареєю, кардитом, асептичним менінгітом. Ураження нирок обумовлене ішемією (гостра ниркова недостатність, гемолітико-уремічний синдром) або супроводжується зниженням ШКФ, еритроцитурією, мононуклеарною лейкоцитурією, раннім зниженням питомої ваги сечі.
Лікування хвороби Кавасакі переважно симптоматичне. Загрозливим для стану дитини є розвиток гострої ниркової недостатності, що потребує проведення діалізу.
Синдром Гудпасчера
МКХ-10 М31.8+N08.5
Синдром Гудпасчера (СГ) належить до групи васкулітів, асоційованих з органоспецифічними антитілами. Захворювання виникає в результаті вироблення організмом автоантитіл, головним чином до базальних мембран клубочкових капілярів нирок і альвеол легень, і виявляється клінікою прогресуючого гломерулонефриту в поєднанні з легеневою кровотечею (кровохарканням).
У світовій літературі існує багато синонімів СГ, наприклад сегментарний некротичний гломеруліт із геморагічним альвеолітом (пневмонітом), легенева пурпура й нефрит, геморагічний пульморенальний синдром, легеневий гемосидероз із нефритом, геморагічна інтерстиціальна пневмонія з нефритом, персистуючий гемофтиз із гломерулонефритом та ін.
Уперше синдром був описаний у 1919 р. гарвардським патологом Е. Гудпасчером, що спостерігав у 18-літнього юнака, який переніс інфлюенцу, анемію на тлі рецидивуючого кровохаркання і двосторонніх легеневих інфільтратів. Після смерті, що настала через 6 тижнів від початку захворювання, на розтині були виявлені альвеолярна геморагія, дифузний некроз альвеол і проліферативний нефрит. Наступні дані про синдром Гудпасчера належать уже до 1955 р., коли Т. Parkin із співавт. описали сім нових випадків поєднання легенево-ниркових уражень, що за клінічною і морфологічною картиною аналогічні описаному Гудпасчером спостереженню. Відтоді кількість нової інформації про дану патологію збільшується з кожним роком.
У 1958 р. М. Stanton і J. Tange повідомили про дев’ять випадків поєднання ураження легень і нирок, що характеризувалися рецидивуючими легеневими кровотечами, гемосидерозом легень і гломерулонефритом, із летальним кінцем через кілька місяців від початку захворювання. Вони вперше описали цей симптомокомплекс і ввели термін «синдром Гудпасчера».
Велику роль у вивченні цього синдрому відіграли численні дослідження, що доводять споріднення антигенних властивостей базальних мембран клубочків нирок і капілярів строми легень.
Критерії встановлення діагнозу синдрому Гудпасчера: кровохаркання, анемія, не пов’язана з тяжкістю ниркової недостатності, швидко прогресуючий гломерулонефрит, наявність автоантитіл до базальної мембрани клубочків (альвеол). У більшості пацієнтів розвивається діалізна ХНН (рис. 4).
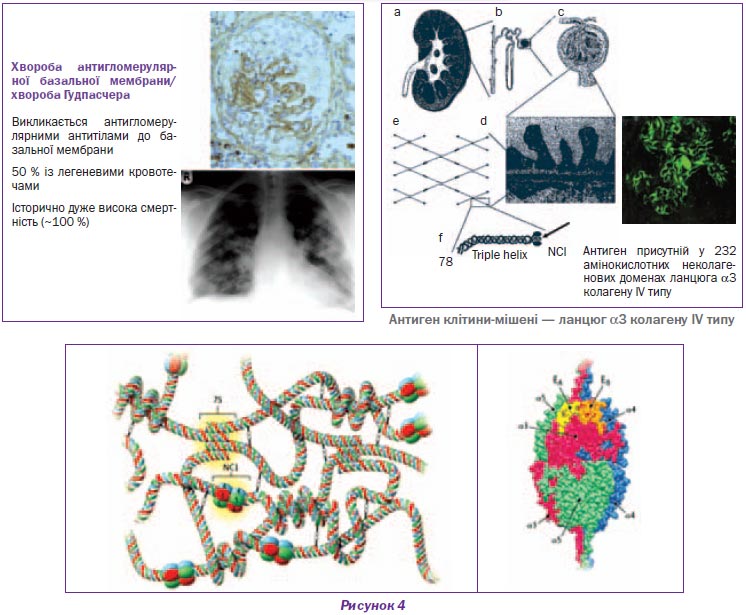
Морфологічні зміни при синдромі Гудпасчера представлені фокальним нефротизуючим ГН і дифузним ГН із півмісяцями.
Приклад діагнозу:синдром Гудпасчера, дифузний ГН із півмісяцями, артеріальна гіпертензія 2-ї ст., анемія 1-го ст.
Лікування синдрому Гудпасчера складається з індукції ремісії й підтримуючої терапії. Для індукції ремісії використовується пульс-терапія преднізолоном (метипредом) 3–5 уведень з одночасним проведеним плазмаферезом 60 мл/кг (максимально до 4 л) протягом перших 2 тижнів до повної елімінації автоантитіл і призначення циклофосфаміду (2–3 мг/кг/добу або 2 мг/кг/добу для осіб віком понад 55 років або у вигляді пульс-терапії). Рання повна елімінація автоантитіл дозволяє домогтися ремісії СГ. Підтримуюча терапія преднізолоном і циклофосфаном дозволяє зменшити ризик рецидиву захворювання. Рання двостороння нефректомія вважається невиправданою. Трансплантація нирки ефективна.
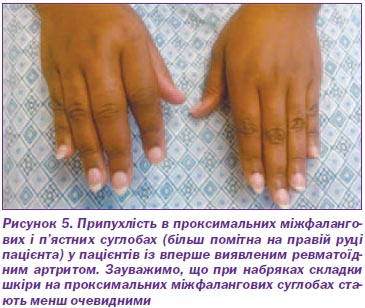
Ураження нирок при ревматичних артритах
До уражень нирок при ревматичних артритах відносяться нефропатії при ревматоїдному артриті, подагрі, псоріатичному артриті та анкілозуючому спондилоартриті.
Ревматоїдний артрит
У грудні 2011 AFP оновив дані про сучасну діагностику та лікування ревматоїдного артриту (http:www.aafp.org/afpsort.xml). Основні положення згідно з доказовою медициною такі:
Пацієнтів із запальними захворюваннями суглобів слід спрямовувати до ревматолога, особливо якщо симптоми тривають понад шість тижнів (С).
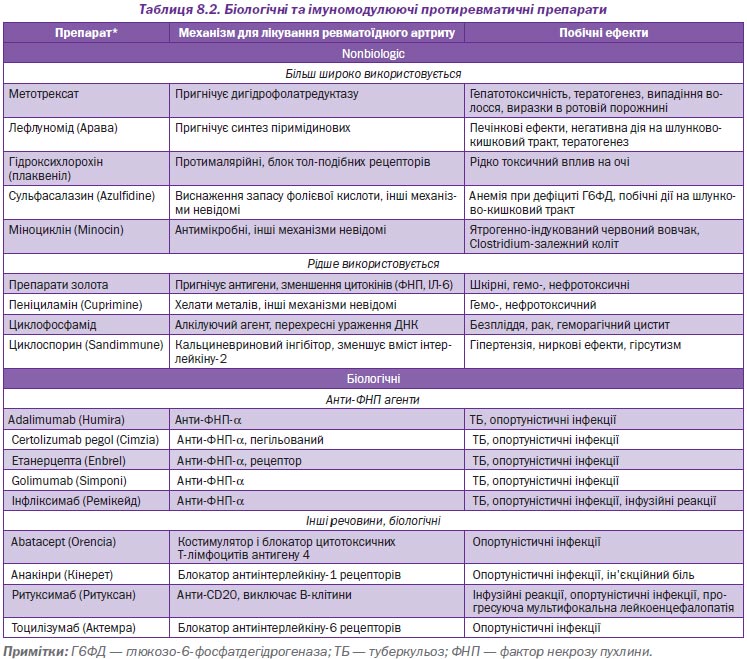
У осіб з РА комбінована терапія двома або більше імуномодулюючими протиревматичними препаратами більш ефективна, ніж монотерапія. Проте не треба використовувати одночасно більше ніж один біологічний агент (наприклад, адалімумаб (Humira) з абатацептом (Orencia)) через високий ризик побічних ефектів (А).
Фізичні вправи можуть поліпшити якість життя та силу м’язів у пацієнтів із РА (В).
Серцево-судинні захворювання є основною причиною смертності в осіб із РА, отже, фактори ризику розвитку ішемічної хвороби серця слід виявляти та модифікувати в таких пацієнтів (С).
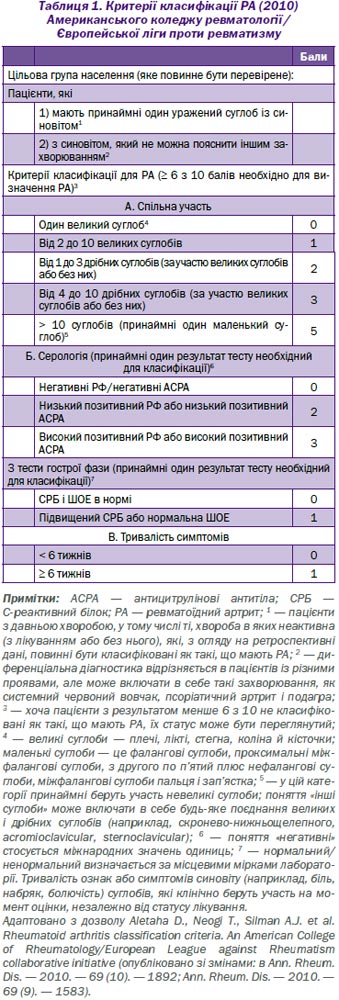
Ревматоїдний артрит є найбільш частим діагнозом системного запального артриту. Жінки, які палять, і ті, які мають сімейну історію цього захворювання, найчастіше страждають від РА. Критерії діагностики включають принаймні один збільшений суглоб, що не пояснюється іншим захворюванням. Ймовірність діагнозу ревматоїдного артриту зростає зі збільшенням ураження числа дрібних суглобів. У пацієнтів із запальним артритом наявність ревматоїдного фактора, або антицитрулінового антитіла, або підвищений рівень С-реактивного білка, або швидкість осідання еритроцитів передбачає діагноз ревматоїдного артриту. Первинна оцінка лабораторних даних повинна також включати повний аналіз крові з диференціальною оцінкою функцій нирок і печінки. Пацієнти, які приймають біологічні агенти, повинні бути перевірені на гепатит В, гепатит С і туберкульоз. Рання діагностика ревматоїдного артриту дозволяє раніше почати лікування з імуномодулюючими протиревматичними засобами. Комбінації препаратів часто використовуються для боротьби з цією хворобою. Метотрексат, як правило, є препаратом першої лінії для лікування ревматоїдного артриту. Біологічні агенти, такі як інгібітори фактора некрозу пухлин, як правило, вважаються агентами другої лінії або можуть бути додані для подвійної терапії. Цілі лікування включають мінімізацію болю в суглобах і набряку, профілактику рентгенографічних ушкоджень і видимих деформацій, а також продовження роботи й особистої діяльності пацієнта.
У групі хворих із системними проявами ревматоїдного артриту ураження нирок відзначене в 1/3 випадків. Клінічно воно відзначається в 10 % хворих і розцінюється як один із найбільш тяжких вісцеритів, нерідко визначає тяжкість перебігу і прогноз. Виділяють три основні групи ураження нирок при РА.
Перша — це амілоїдоз нирок, що нерідко призводить до ниркової недостатності. Іноді він зустрічається в ранній термін захворювання (через 1–2 роки після початку РА), розвивається відповідно до активності процесу, виникає більш гостро у хворих із максимальними імунологічними порушеннями. З прогресуванням амілоїдозу згладжуються суглобові прояви і на перший план виступає нирково-уремічний синдром. Однак частіше амілоїдоз при РА розвивається поступово, а іноді може виявитися й у більш віддаленому періоді, через 15–25 років. Важлива ознака амілоїдозу нирок — протеїнурія при невеликому сечовому осаді. Кількість білка коливається від 0,033 до 10–15 г/л і більше. Поступово розвивається гіпопротеїнемія, з’являються набряки, гіперхолестеринемія, тобто ознаки нефротичного синдрому з подальшим розвитком ниркової недостатності, можливо, і з артеріальною гіпертензією. Амілоїдоз при РА майже завжди має системний характер та супроводжується збільшенням печінки, селезінки, лімфатичних вузлів і ураженням інших органів. Зрідка відкладення амілоїду «неактивні», тобто клінічні прояви не міняються протягом багатьох років.
Друга група ураження нирок при РА — гломерулонефрит. Гломерулонефрит виявляється значно рідше, ніж ревматоїдний амілоїдоз, що пояснюється особливостями циркулюючих у крові імунних комплексів. Розвивається гломерулонефрит на початку хвороби при бурхливому перебігу ревматоїдного артриту. Морфологічно це мембранозний або мембранозно-проліферативний осередковий або дифузний гломерулонефрит. Етіологічним фактором у розвитку гломерулонефриту також можуть бути солі золота й Д-пеніциламін. Клінічно він може виражатися минущою протеїнурією, аж до розвитку нефротичного синдрому, що спостерігається в 30 % хворих. При лікуванні вольтареном можливий розвиток екстрамембранозного гломерулонефриту з нефротичним синдромом. Після відміни препарату нерідко відбувається повна ремісія зі зникненням всіх ознак нефротичного синдрому без додаткової терапії.
Третя група — це інтерстиціальне ураження нирок, обумовлене не самим РА, а тривалим прийомом пацієнтом нестероїдних протизапальних препаратів у зв’язку з ревматоїдним артритом. Сосочковий некроз і пієлонефрит часто обумовлені лікарською терапією. Деякі протиревматичні препарати викликають зміни в нирках (папілярний некроз у хворих, які приймали фенацетин, «золота» нефропатія з ушкодженням канальців і клубочків, нефропатії при лікуванні Д-пеніциламіном та ін.). Можливий розвиток некротичного папіляриту при лікуванні нестероїдними протизапальними препаратами. Для діагностики сосочкового некрозу використовується комп’ютерна томографія.
Подагра
Подагра — це захворювання, пов’язане з порушенням метаболізму пуринів і накопиченням підвищеної кількості сечової кислоти в організмі, а також із відкладенням і накопиченням уратів у тканинах опорно-рухового апарату і внутрішніх органів. Для хвороби характерні гіперурикемія, подагричний артрит, ураження нирок за типом інтерстиціального нефриту й уролітіаз.
Захворювання пов’язане як із деякими генетичними дефектами, що впливають тією або іншою мірою на механізм пуринового обміну, так і з дією аліментарних факторів.
Гіперурикемія і подагра — поняття неоднозначні. Гіперурикемія трансформується в подагру, якщо виникають умови для кристалізації уратів і лейкоцитарної інфільтрації зон-мішеней. Нижчий поріг урикемії, при якому не було б ризику виникнення подагри, не визначений, а от висока гіперурикемія є чинником ризику. Подагра звичайно починається у віці 25–60 років, уражає переважно чоловіків, жінки хворіють рідше, у переважній більшості випадків у період менопаузи. Іноді виділяють подагру без гіперурикемії, але з підвищеним вмістом моноурату натрію.
Виділяють також вторинну подагру при захворюваннях крові, при прийомі цитостатиків, променевій терапії злоякісних новоутворень. Вторинна подагра може виникнути і при хронічній нирковій недостатності у зв’язку з втратою нирками здатності до достатнього виведення уратів. Вторинна гіперурикемія може бути викликана тіазидними діуретиками, саліцилатами.
Клініка інтерстиціального нефриту не має яскраво виражених особливостей. Сечовий синдром характеризується невеликою (не більше 1,5 г/л), але постійною протеїнурією, незначною гематурією і лейкоцитурією, зниженням відносної щільності сечі. Такого роду зміни в сечі найчастіше змушують думати про хронічний дифузний гломерулонефрит з ізольованим сечовим синдромом, і якщо в хворого в анамнезі відсутнє ураження суглобів (класичні подагричні кризи або атипові форми), діагностика досить складна. До деякої міри орієнтирами повинні служити такі дані: чоловіча стать (вік 30–50 років), нормо- або гіперстенічна статура, наявність тофусів на вушних раковинах.
В усіх випадках, коли виникає думка про подагричний нефрит, вирішальним діагностичним критерієм є концентрація сечової кислоти в крові. Це дуже важливо, оскільки відомо, що нефропатія часто передує ураженню суглобів іноді на кілька років, а призначення в таких випадках алопуринолу дозволяє зменшити сечовий синдром і запобігти розвитку уролітіазу й подагричного артриту.
Другим за частотою типом ураження є уролітіаз, що характеризується нирковими кольками й відходженням конкрементів (іноді великої кількості протягом багатьох років), що є рентгенонегативними, тому що представлені переважно сечовою кислотою. Дуже рідко зустрічається гостра анурія з ГНН, пов’язана з випаданням великої кількості кристалів сечової кислоти в канальцях. Така патологія відзначається при вторинній подагрі, тобто у хворих із лейкозами й іншими лімфопроліферативними захворюваннями на тлі масивної терапії цитостатиками й пов’язаним із цим швидким руйнуванням багатьох клітин із виділенням величезної кількості нуклеопротеїдів (tumor lysis syndrome). У цьому випадку призначається внутрішньовенний препарат фастуртек, що повністю інгібує утворення сечової кислоти. Крім того, можливо гостре подагричне запалення нирок, що перебігає з гострими болями в попереку, лихоманкою, гематурією, які зникають після проведення антиподагричної терапії. Інтерстиціальний подагричний нефрит та уролітіаз часто ускладнюються пієлонефритом, розвитком артеріальної гіпертензії й у багатьох випадках призводять до смерті від хронічної ниркової недостатності.
Диференціальна діагностика повинна проводитися в першу чергу з хронічним гломерулонефритом, а у випадку уратного нефролітіазу обов’язкове уточнення анамнезу (суглобові кризи) і визначення рівня сечової кислоти в крові для встановлення його можливого подагричного генезу.
Подагричну нефропатію лікують як подагру взагалі і не включають лише засоби, застосування яких націлене на ліквідацію гострих суглобових кризів. Лікування повинне бути комплексним, впливати на патогенетичні фактори захворювання й різні його симптоми.
Рекомендується дієта з різким обмеженням продуктів, що містять пурини у великій кількості. До них відносяться печінка, нирки, мозок, м’ясо, гриби, м’ясні бульйони, холодець, ковбаси, шинка, риба, квасоля, боби, сочевиця, какао, міцний чай і кава, шоколад. Шкідливий прийом алкоголю, тому що спирт зменшує виведення сечової кислоти нирками.
З огляду на те що подагричну нефропатію нерідко супроводжує оксалатурія (частіше в жінок), не рекомендується вживати щавель, шпинат, селеру, перець, редис, брукву. Необхідно багато пити (не менше 2 л у добу) — лужні мінеральні води, молоко, фруктові й овочеві соки. Відварне м’ясо дозволяється по 60–100 г 2 рази на тиждень; без обмежень — сир, сметана, молоко, кисломолочні продукти, яйця, овочі, фрукти, вироби з круп, макарони; із приправ і спецій — оцет, лавровий лист, лимони. Бідна пуринами дієта може допомогти знизити урикемію не більше ніж на 0,12 ммоль/л, чого, однак, цілком достатньо при помірної гіперурикемії, а в більш тяжких випадках вона дозволяє обмежити прийом медикаментозних засобів.
Медикаменти, застосовувані для лікування хронічної подагри, а отже і подагричної нефропатії, поділяються на дві групи: урикозуричні й урикодепресорні. Застосування тих або інших залежить від ступеня урикозурії. Якщо нирки виводять менше 600 мг сечової кислоти на добу, подагра розцінюється переважно як ниркова й лікувати її потрібно урикозуричними препаратами, а якщо при гиперурикемії визначається ще й урикозурія (більше 600 мг на добу), то подагра розцінюється як метаболічна, тоді варто призначати препарати, що справляють урикодепресорний ефект.
До урикозуричних препаратів відносять етамід, антуран і їх аналоги, до урикодепресивних — алопуринол і оротову кислоту. Алопуринол ефективний при лікуванні усіх форм подагри, тому що здатен істотно знижувати урикемію й одночасно трохи збільшувати урикозурію. Він має найменше число побічних дій з усіх препаратів і придатний для тривалого лікування, у тому числі й підтримуючого. На початку лікування призначають по 0,3 г/добу вранці після сніданку, потім, орієнтуючись на зниження урикемії, дають підтримуючі дози 0,1–0,2 г/добу. У пацієнтів, які не піддаються лікуванню алопуринолом, використовують фенофібрат й ентеросорбенти. Дуже ефективним є застосування замість алопуринолу фебукостату, більш селективного за своєю дією, у дозі 40–120 мг/добу.
Для купірування ниркової кольки призначають фітопрепарати — уролесан, спазмоцистенал, канефрон Н. Обов’язковою є корекція рН сечі за допомогою цитратних сумішей, блемарену, магурліту, ураліту.
Застосування будь-яких препаратів, здатних підсилити виділення сечової кислоти нирками (особливо у великих дозах), вимагає постійного контролю за ступенем урикозурії, що не повинна перевищувати 1000 мг на добу, а добовий діурез повинен бути не менше 2 л. В іншому випадку досить реальний ризик загострення нефропатії, оскільки всі урикозуричні препарати можуть витісняти солі сечової кислоти з її відкладень у тканинах і сприяти накопиченню вільних уратів у плазмі. При ХНН і вторинній подагрі алопуринол є препаратом вибору, його добова доза становить 0,1 г.
Через часту гіпертензію, що розвивається, а також лікування протеїнурії й інтерстиціального ураження нирок пацієнтам призначають ІАПФ і сартани. При цьому віддається перевага препаратам із позанирковим шляхом виведення: серед ІАПФ — моексиприлу, моноприлу, серед сартанів — мікардису й теветену. Сартани й особливо ІАПФ із нирковим шляхом виведення сповільнюють виведення сечової кислоти і провокують загострення подагри. Більше того, у пацієнтів із субклінічним зниженням функції нирок (3–4-ї стадії ХХН, цукровим діабетом 2-го типу, метаболічним синдромом) призначення кандесартану, лозартану, периндоприлу, лізиноприлу може призводити до гіперурикемії з відкладенням солей сечової кислоти в дрібних суглобах кистей рук. Особливо чітко цей ефект виявляється в пацієнтів, що у харчовому раціоні віддають перевагу м’ясним продуктам або мають супутні хронічні захворювання печінки. Заміна препарату на одногрупний, але з позанирковим шляхом виведення, сприяє ліквідації артриту.
Необхідно дотримуватися дієти при спадковій обтяженості. Профілактика ураження нирок при наявності в анамнезі суглобового подагричного артриту зводиться до правильного тривалого лікування, спрямованого на нормалізацію рівня сечової кислоти в крові.
Хворі з подагричною нефропатією підлягають диспансеризації, аналогічній такій при хронічному гломерулонефриті, за умови обов’язкового визначення рівня урикемії 1 раз на 2–3 місяці для корекції підтримуючої терапії.
Ураження нирок, обумовлені системними захворюваннями сполучної тканини
До уражень нирок, які виникають як прояви системних захворювань сполучної тканини, відносяться нефрити при СЧВ (розглянуто в розділі дрібносудинних васкулітів), системній склеродермії, дерматоміозиті, синдромі Шегрена, синдромі Шарпа, антифосфоліпідному синдромі та мієломній хворобі.
Системна склеродермія
Для цього захворювання (частіше на нього хворіють жінки) характерні прогресуючі фіброзно-склеротичні зміни в шкірі, підшкірній клітковині та внутрішніх органах, а також генералізоване ураження судин мікроциркуляторного русла.
Згідно з Американським коледжем ревматології (C.W. Brown, 1999) виділяють такі діагностичні критерії системної склеродермії:
Великий критерій — проксимальний або дифузний склероз.
Малі критерії:
- склеродактилія;
- поглиблення на пальцях, фляки або втрата тканини в ділянках подушечок пальців;
- двосторонній фіброз нижніх відділів легень.
Для встановлення діагнозу потрібна наявність одного великого і двох малих критеріїв.
Найбільш типові ознаки склеродермії — ураження шкіри, синдром Рейно. Одночасно спостерігаються зміни в легенях, серці, шлунково-кишковому тракті, кістках (остеоліз нігтьових фаланг), суглобах, печінці, селезінці. Можна відзначити деяку своєрідність ураження нирок при системній склеродермії: зміну міжчасточкових артерій із мукоїдним набряком інтими, звуження їх просвіту, що відбиває загальний генералізований характер ураження судин мікроциркуляторного русла.
У початковій стадії захворювання клінічні й функціональні дослідження нирок малоінформативні, оскільки клубочкова фільтрація і канальцева реабсорбція довго залишаються нормальними. При тривалому перебігу захворювання за допомогою радіонуклідної реносцинтиграфії можна встановити зниження ниркового кровотоку. Гарні результати в розпізнаванні системної склеродермії дає також біохімічна діагностика ураження нирок: виявляється підвищена екскреція з сечею кислих фосфатаз і серомукоїдних білків.
Клініка ураження нирок при системній склеродермії повністю визначається характером патологічного процесу, що розвивається в них. Розрізняють три основні варіанти його перебігу: гострий (зустрічається порівняно нечасто), підгострий і хронічний. Найбільш тяжким ураженням при гострому перебігу є розвиток так званої справжньої склеродермічної нирки, коли внаслідок кортикальних некрозів через генералізоване ураження судин раптово починається й бурхливо прогресує гостра ниркова недостатність з артеріальною гіпертензією, ретино- й енцефалопатією. Зазначене ускладнення виявляється на першому-другому році захворювання. Спочатку констатують помірну протеїнурію й невеликі зміни в осаді сечі. Надалі, протягом 1–2 тижнів, протеїнурія наростає, порушується функція нирок і у 2/3 хворих відзначається злоякісна артеріальна гіпертензія. Швидко розвиваються олігурія, анурія, азотемія, що нерідко призводить до летального кінця.
При підгострому перебігу системної склеродермії ураження нирок більш доброякісне. Воно перебігає за типом інтерстиціального нефриту або хронічного гломерулонефриту з гіпертензивним синдромом і згодом із порушенням азотовидільної функції. Нефротичний синдром при системній склеродермії зустрічається вкрай рідко.
В 1/3 хворих відзначається ураження нирок за типом латентного нефриту або субклінічної форми нефропатії з переважними функціональними порушеннями. Для цього випадку характерні незначні зміни сечового осаду й протеїнурія. Захворювання прогресує у ниркову недостатність. Частіше мінімальні ураження нирок без ознак прогресування спостерігаються роками. Хворі цієї групи помирають від інших причин. Однак на розтині виявляються зміни, що відповідають картині мембранозного гломерулонефриту з наявністю судинних і стромальних змін або проміжного й периваскулярного нефросклерозу.
Лікування справжньої склеродермічної нирки спрямоване на боротьбу з гострою нирковою недостатністю й гіпертонічною енцефалопатією (гепарини, корекція водно-сольового обміну, осмотичні діуретики, великі дози лазиксу, судинорозширювальні препарати, потужні гіпотензивні засоби). Щодо останніх необхідно підкреслити, що, за сучасними даними, інгібітори АПФ/сартани є препаратами вибору при розвитку гострої склеродермічної нирки й ренінзалежної гіпертензії у хворих із системною склеродермією. З огляду на тяжкість перебігу і швидкопрогресуючий характер гострої склеродермічної нефропатії їх варто комбінувати з фізіотензом й окремими антагоністами кальцію (фелодипін, лерканідипін, дилтіазем).
Протипоказане застосування антибіотиків, сульфаніламідних, білкових препаратів, небажане використання кортикостероїдів.
При усіх формах системної склеродермії, особливо при тяжкому прогресуючому її перебігу, найбільш обнадійливі результати спостерігаються при лікуванні моноклональними антитілами з використанням сучасних імуносупресивних препаратів. Із традиційних відомих засобів використовують Д-пеніциламін (купреніл), що перешкоджає утворенню поперечних зв’язків колагену й переходу розчинної його фракції в нерозчинну. Крім того, Д-пеніциламін може перетворювати вже сформований нерозчинний колаген у розчинну форму. Лікування починають у стаціонарі. Первинна доза — 300 мг на добу всередину в капсулах (капсула — 150 мг). Поступово до кінця третього тижня дозу збільшують до 600–900 мг на добу, через місяць її знижують і до кінця третього місяця доводять до підтримуючої — 150 мг на добу. Необхідний постійний клініко-лабораторний контроль. З огляду на високу токсичність купренілу останній нерідко заміняють унітіолом у поєднанні з ІАПФ/сартанами й тиклопедином.
При значній активності процесу, гострому і підгострому його перебігу разом із купренілом показані низькі дози кортикостероїдів. Призначають 20–40 мг преднізолону на добу з поступовим зниженням дози в міру зменшення активності процесу. Застосування підтримуючих доз преднізолону може бути більш тривалим.
За відсутності ефекту від Д-пеніциламіну або при його непереносимості, а також при високій активності склеродермічного процесу, що не купірується кортикостероїдами, або при виникненні протипоказань до них призначають імуносупресори (азатіоприн та ін.). У зазначених ситуаціях можуть бути успішно використані й методи екстракорпорального очищення крові — імуносорбція і плазмаферез.
При розвитку термінальної ХПН показані діаліз і трансплантація нирки.
Дерматоміозит
Ідіопатичний дерматоміозит починається поступово, частіше зустрічається в жінок, ніж у чоловіків (2 : 1). Хвороба виявляється ураженням шкірних покривів (параорбітальний набряк і темно-лілова еритема, капілярити долонь) або м’язовим синдромом із прогресуючою м’язовою слабкістю і вираженим симетричним ураженням насамперед проксимальних м’язів кінцівок. Захворювання перебігає за змішаним типом зі шкірно-м’язовими проявами. Будь-який із варіантів початку дерматоміозиту розвивається з такими загальними симптомами, як субфебрильна температура, прогресуюча втрата маси тіла, трофічні зміни шкірних покривів. Можливі й вісцеральні прояви, що звичайно не настільки виражені, як при системному червоному вовчаку й системній склеродермії.
Ураження нирок при дерматоміозиті різноманітне. Найбільш частим є розвиток інтерстиціального нефриту й гломерулонефриту, що характеризуються підгострим або хронічним перебігом із різними клінічними варіантами й наслідками (гіпертензивний синдром, ниркова недостатність). При гострому й підгострому перебігу дерматоміозиту з генералізацією процесу іноді розвивається тяжкий нирковий васкуліт із реноваскулярною гіпертензією й ретинопатією. Ураження нирок можливе й за рахунок сучасної активної терапії (антибактеріальної, нестероїдної, глюкокортикоїдної). Зрідка сечовий синдром при дерматоміозиті обумовлений міоглобінурією, що виявляється темно-коричневим кольором сечі. У сироватці крові при цьому виявляють високу активність ферментів.
Діагностика дерматоміозиту ґрунтується переважно на клінічній картині. Дані біопсії м’язів (бліді, «варені» м’язи з некрозом м’язових волокон, периваскулярною інфільтрацією й атрофією волокон) та електроміографії (низькоамплітудна електрична активність) неспецифічні і, отже, великої діагностичної цінності не мають.
До 23 % випадків дерматоміозиту обумовлені неопластичним процесом (утворення злоякісних пухлин різної локалізації). Клініка паранеопластичного дерматоміозиту ідентична клініці ідіопатичного. Прогноз його залежить від лікування основного захворювання. При паранеопластичному дерматоміозиті й успішному видаленні пухлини зникають всі ознаки хвороби.
Лікування ідіопатичного дерматоміозиту включає можливість проведення пульс-терапії глюкокортикоїдами й цитостатиками з одночасною синхронізацією з плазмаферезом або імуносорбцією. Частіше застосовуються невисокі дози глюкокортикоїдів (до 60 мг на добу). Приймають їх протягом 2–3 місяців і більше, потім дозу поступово знижують і доводять до підтримуючої. У період клінічної ремісії можлива повна відміна кортикостероїдів. Нерідко замість цитостатиків призначають амінохінолінові препарати (плаквеніл, делагіл), що рекомендують приймати довгостроково, протягом декількох років. Обов’язковими патогенетичними препаратами є ІАПФ і сартани в пожиттєвому призначенні.
Антифосфоліпідний синдром
Антифосфоліпідний синдром (АФС) належить до числа найбільш актуальних мультидисциплінарних проблем сучасної медицини й розглядається як унікальна модель автоімунної тромботичної васкулопатії.
Початок вивченню АФС був покладений близько ста років тому в працях A. Wassermann, присвячених лабораторному методу діагностики сифілісу. При проведенні скринінгових досліджень стало очевидним, що позитивну реакцію Вассермана можливо виявити в багатьох людей без клінічних ознак сифілітичної інфекції. Цей феномен отримав назву «біологічна хибнопозитивна реакція Вассермана». Далі було встановлено, що основним антигенним компонентом в реакції Вассермана є негативно заряджений фосфоліпід, названий кардіоліпіном. Упровадження радіоімунологічного, а потім й імуноферментного методу (ІФМ) визначення антитіл до кардіоліпінів (аКЛ) сприяло більш глибокому розумінню їх ролі при захворюваннях людини. За сучасними уявленнями, антифосфоліпідні антитіла (аФЛ) — це гетерогенна популяція автоантитіл, що взаємодіють із негативно зарядженими, рідше нейтральними фосфоліпідами і/або фосфоліпідзв’язуючими сироватковими білками. Залежно від методу визначення аФЛ умовно поділяють на три групи: ті, що виявляють за допомогою ІФМ із використанням кардіоліпіну, рідше інших фосфоліпідів; антитіла, що виявляють за допомогою функціональних тестів (вовчаковий антикоагулянт); антитіла, що не діагностуються за допомогою стандартних методів (антитіла до білка С, S, тромбомодуліну, гепарансульфату, ендотелію та ін.).
Наслідком великої зацікавленості у вивченні ролі аФЛ та вдосконаленні методів лабораторної діагностики став висновок, що аФЛ є серологічним маркером симптомокомплексу, який включає венозні і/або артеріальні тромбози, різні форми акушерської патології, тромбоцитопенію, а також широкий спектр неврологічних, шкірних, серцево-судинних порушень. Починаючи з 1986 р. цей симптомокомплекс став визначатися як антифосфоліпідний синдром, а в 1994 р. на міжнародному симпозіумі з аФЛ було запропоновано також використовувати термін «синдром Hughes» — за ім’ям англійського ревматолога, який зробив найбільший внесок у вивчення цієї проблеми.
Справжня поширеність АФС у популяції досі невідома. Оскільки синтез аФЛ можливий і в нормі, низький рівень антитіл нерідко зустрічається в крові здорових людей. За різними даними, частота виявлення аКЛ у популяції коливається від 0 до 14 %, у середньому ж вона становить 2–4 %, при цьому високі титри виявляють достатньо рідко — приблизно в 0,2 % донорів. Дещо частіше аФЛ зустрічається в осіб похилого віку. При цьому клінічне значення аФЛ у здорових осіб (які не мають явних симптомів захворювання) не зовсім ясне. Часто при повторних аналізах рівень підвищених у попередніх аналізах антитіл нормалізується.
Наростання частоти зустрічальності аФЛ відзначене при декількох запальних, автоімунних й інфекційних захворюваннях, злоякісних новоутвореннях, на фоні прийому лікарських препаратів (оральні контрацептиви, психотропні засоби та ін.). Є дані щодо імуногенетичної схильності до підвищеного синтезу аФЛ і більш частого їх виявлення в родичів хворих з АФС.
Доведено, що аФЛ не тільки серологічний маркер, але й важливий патогенетичний медіатор, що викликає розвиток основних клінічних проявів АФС. Антифосфоліпідні антитіла мають здатність діяти на більшість процесів, становлячи основу регуляції гемостазу, порушення яких призводить до гіперкоагуляції. Клінічне значення аФЛ залежить від того, чи пов’язана їх наявність у сироватці крові з розвитком характерної симптоматики. Так, прояви АФС відзначаються тільки в 30 % хворих із позитивним вовчаковим антикоагулянтом і в 30–50 % хворих, які мають помірний або високий рівень аКЛ. Захворювання розвивається переважно в молодому віці, при цьому АФС може бути діагностованим у дітей і навіть у новонароджених. Як і інші автоімунні ревматичні захворювання, даний симптомокомплекс частіше зустрічається в жінок, ніж у чоловіків (співвідношення 5 : 1).
Найбільш частими й характерними проявами АФС є венозні і/або артеріальні тромбози й акушерська патологія. При АФС можуть вражатися судини будь-якого калібру й локалізації — від капілярів до великих венозних та артеріальних стовбурів. Тому спектр клінічних проявів дуже різноманітний і залежить від локалізації тромбозу. За сучасними уявленнями, основу АФС становить своєрідна васкулопатія, обумовлена незапальним і/або тромботичним ураженням судин, що завершується їх оклюзією. У рамках АФС описані патологія ЦНС, серцево-судинної системи, порушення функції нирок, печінки, ендокринних органів, шлунково-кишкового тракту. Із тромбозом судин плаценти пов’язують розвиток деяких форм акушерської патології.
Ниркова патологія достатньо різноманітна при АФС. У більшості пацієнтів спостерігається тільки безсимптомна помірна протеїнурія (менше 2 г на добу), без порушення функції нирок, але може розвиватися гостра ниркова недостатність із вираженою протеїнурією (аж до нефротичного синдрому), активним сечовим осадом і артеріальною гіпертензією. Ураження нирок пов’язане головним чином із внутрішньоклубочковим мікротромбозом і визначається як ниркова тромботична мікроангіопатія.
Ведення хворих з АФС (як і з іншими тромбофіліями) базується на призначенні антикоагулянтів непрямої дії (варфарин, аценокумарол) та антиагрегантів (у першу чергу низьких доз ацетилсаліцилової кислоти — АСК). Це пов’язано перш за все з тим, що для АФС характерний високий ризик повторних тромбозів, що значно переважає такий при ідіопатичних венозних тромбозах. Припускають, що більшість хворих з АФС із тромбозами потребує профілактичної антиагрегантної і/або антикоагулянтної терапії протягом тривалого часу, а іноді й усього життя. Крім того, ризик первиних і повторних тромбозів при АФС необхідно знижувати шляхом впливу на такі кориговані фактори ризику, як гіперліпідемія (статини: симвастин — симвастол, симло; ловастатин — ровакор, кардіостатин; правастатин — ліпостат; аторвастатин — авас, ліпримар; фібрати: безафібрат — холестенорм; фенофібрат — нофібал, грофібрат; ципрофібрат — ліпанор), артеріальна гіпертензія (інгібітори АПФ — капотен, синоприл, диротон, моекс; b-блокатори — атенолол, конкор, егілок, беталок ЗОК, дилатренд; антагоністи кальцію — амловас, норваск, нормодипін, лацидипін), гіпергомоцистеїнемія, малорухомий спосіб життя, паління, прийом оральних контрацептивів та ін.
В останні роки інтенсивно розробляють нові антитромботичні агенти, до яких зараховують гепариноїди (гепароїд лечива, емеран, сулодексид — вессел дуе), інгібітори тромбоцитарних рецепторів (тиклопідин, тагрен, клопідогрель) та інші препарати. Попередні клінічні дані свідчать про безсумнівну перспективність цих лікарських засобів.
Усі хворі з АФС повинні знаходитися під тривалим диспансерним наглядом, першочерговим завданням якого є оцінка ризику рецидивів тромбозів та їх профілактика. Необхідний контроль активності основного захворювання (при вторинному АФС), своєчасне виявлення й лікування супутньої патології, у тому числі інфекційних ускладнень, а також вплив на кориговані фактори ризику тромбозів. Установлено, що прогностично несприятливими факторами щодо летальності при АФС є артеріальні тромбози, висока частота тромботичних ускладнень і тромбоцитопенія, а з лабораторних маркерів — наявність вовчакового антикоагулянту. Перебіг АФС, тяжкість і поширеність тромботичних ускладнень непередбачувані; універсальні схеми терапії, на жаль, відсутні. Вищевказані факти, а також поліорганність симптоматики потребують об’єднання лікарів різних фахів для вирішення проблем, пов’язаних із веденням даної категорії хворих.
Ураження нирок при ревматизмі
Ревматичний процес приблизно в 5 % випадків призводить до ураження нирок. Основа ниркової патології при ревматизмі — васкуліти, що при тяжкому перебігу хвороби схильні до генералізації. При активній фазі ревматизму в нирках відбуваються запальні зміни в судинах із підвищенням їх проникності. Особливо тяжкі зміни виявляються в часточкових артеріях і приносних судинах клубочків. При неактивній фазі ревматизму в цих судинах переважають склеротичні зміни, ступінь вираженості яких залежить від перенесених ревматичних атак.
Найбільш частою патологією нирок при ревматичній лихоманці є ізольований сечовий синдром. У той же час у період стрептококової інфікованості й сенсибілізації може відзначатися гостре інфекційне ураження за типом минущого інфекційного нефриту (інфекційна нирка за Е.М. Тареєвим).
Симптоми ураження нирок частіше виникають при повторних атаках ревматизму. У хворих із затяжним й, особливо, постійно рецидивуючим перебігом ревматизму частіше зустрічається латентний нефрит, що виявляється помірним, але стійким сечовим синдромом (стійка протеїнурія, мікрогематурія) при відсутності скарг.
Ревматичний дифузний гломерулонефрит зустрічається рідко і становить 0,67–1 % серед хворих на гломерулонефрит. При розвитку його можуть спостерігатися різні варіанти перебігу: латентний, гіпертензивний, нефротичний, змішаний. Поступово знижується концентраційна функція нирок. Можливий розвиток нефросклерозу. Набряки у хворих на ревматизм із пороком серця, що сформувався, важкі для діагностики через наявність серцевої недостатності.
Найчастіше доводиться диференціювати ревматичний нефрит і «серцеву» застійну нирку, що розвивається в результаті гіпоксії внаслідок гемодинамічних порушень. При застійній нирці характерна еритроцитурія, сечовий синдром нестабільний і зменшується або зникає при лікуванні кардіотонічними й сечогінними засобами.
У хворих на ревматизм при наявності пороку серця бувають інфаркти нирок (на розтині в 1/3 випадків). Як правило, ці інфаркти, навіть множинні, перебігають безсимптомно, характеризуючи «німий» перебіг.
Наявність ниркового синдрому у хворих на ревматизм нерідко обумовлена приєднанням затяжного септичного ендокардиту, що підтверджується виявленням у крові збудника і лікувальним ефектом антибактеріальної терапії.
Спеціальних методів і засобів лікування ревматичного нефриту немає. Як правило, проводиться комплексна терапія ревматичної лихоманки із включенням антибактеріальних засобів, ІАПФ і сартанів.
Ураження нирок при мієломній хворобі
Мієломна хвороба (мієлома, плазмоцитома) — це системне захворювання пухлинно-гіперпластичного типу з переважним ураженням кісток скелета, що характеризується злоякісною проліферацією клітин ретикулоплазматичної природи.
Ураження нирок розцінюється як найбільш частий клінічний, морфологічний і лабораторний (біохімічний) прояв мієломної хвороби й у той же час як одне з найбільш тяжких і несприятливих у прогностичному плані ускладнень цієї хвороби. Частота ураження нирок при мієломній хворобі коливається від 60 до 100 %. У багатьох випадках патологічні зміни в нирках служать першими, найбільш ранніми клініко-лабораторними проявами мієломної хвороби, що стало підставою для виділення ниркової форми цієї хвороби. Ураження нирок, обумовлене мієломною хворобою, визначають як мієломну нефропатію або мієломну нирку, рідше — як парапротеїнемічний нефроз. Патологічні зміни в нирках можуть мати різний характер і відрізнятися значним поліморфізмом. В одних випадках вони строго специфічні для мієломної хвороби й обумовлені пара- і диспротеїнозом. Такому характеру ураження нирок і відповідає термін «мієломна нирка». В інших випадках мієломної нефропатії зміни в нирках мають неспецифічний (або не строго специфічний) для даної хвороби характер і виявляються пієлонефритом, амілоїдозом нирок, нефрокальцинозом, артеріолосклерозом.
Порівняно часті пієлонефрит й артеріолосклероз нирок пояснюють переважанням серед хворих на мієломну хворобу осіб літнього віку і зниженням при цьому захворюванні опірності організму до інфекції.
Патогенез мієломної нефропатії заснований на ушкодженні ниркових нефронів патологічними (аномальними) білками — парапротеїнами. З огляду на це мієломна нефропатія за своїм походженням розглядається як класичний приклад нефриту виділення. Специфічним для справжньої мієломної нирки вважається відкладення преципітатів патологічних мієломних білків у дистальних відділах канальців з ушкодженням останніх. При цьому синтезовані мієломними клітинами мікромолекулярні білки Бенс-Джонса надходять у кровотік, досягають нирок і легко проходять через неушкоджений клубочковий фільтр. У просвіті канальців, де первинна сеча має кислу реакцію, білкові маси, що профільтрувалися, в клубочках згортаються, утворюють велику кількість циліндрів, що призводять до обтурації просвіту дистальних відділів канальців. У результаті підвищується внутрішньоканальцевий тиск у проксимальних відділах канальців із розширенням їх просвіту і розвитком так званого інтраренального гідронефрозу (нефрогідрозу). Крім того, частково реабсорбовані канальцевим епітелієм патологічні білки проникають в інтерстиціальні тканини, викликаючи набряклість ниркової строми, застій лімфи з виникненням лімфоцитарних інфільтратів, тобто запальний процес в інтерстиціальній тканині (інтерстиціальний нефрит). Надалі розвиваються гіаліноз і склероз інтерстицію з подальшою загибеллю клубочків, нефронів і нефротичним зморщенням нирок. Ушкодження канальців може обумовити розвиток нефротичного синдрому.
Всі інші ураження нирок, що виявляються при мієломній хворобі, мають неспецифічний характер. Так, у результаті приєднання інфекції може виникнути пієлонефрит. Приблизно в 5–25 % хворих розвивається амілоїдоз нирок. Метаболічні порушення нерідко призводять до розвитку не тільки інтерстиціального нефриту, але й нефрокальцинозу й уролітіазу.
Клінічна картина мієломної нефропатії доволі різноманітна. Це залежить як від характеру патологічних змін із боку нирок, так і від симптомів ураження інших органів і систем, головним чином кісткової системи. Найбільш ранньою і постійною ознакою мієломної нирки є протеїнурія, що виявляється в 65–100 % хворих. Вираженість її коливається в широких межах — від слідів білка до 3,3–10 г/л. Відомі випадки, коли стійка протеїнурія була єдиним симптомом мієломної хвороби протягом багатьох років. Іноді протеїнурія може задовго передувати появі інших симптомів цієї хвороби. У подібних випадках захворювання довго перебігає під маскою хронічного гломерулонефриту з ізольованим сечовим синдромом. За допомогою електрофорезу білків сечі (з добового її обсягу) на папері або в крохмальному гелі, а також імуноелектро-форезу вдається встановити мікромолекулярну (білок Бенс-Джонса) природу мієломного уропротеїну у вигляді моноклонового піку (М-градієнта) подібно до аналогічного піку на електрофореграмі білків сироватки крові. Уропротеїн Бенс-Джонса електрофоретично виявляється в сечі в 95 % хворих на мієломну хворобу. Тому в кожному випадку протеїнурії неясної етіології, особливо в осіб літнього віку, як правило, необхідно проводити електрофорез білків сечі, тобто досліджувати уропротеїнограму. Для уропротеїнограми хворих на мієломну нефропатію на відміну від протеїнурії іншого походження характерне переважання глобулінів над альбумінами з наявністю піку глобулінурії. З огляду на сказане якісна характеристика білків сечі за допомогою згаданих методів має винятково важливе діагностичне значення.
Еритроцитурія не характерна для мієломної нирки, і лише в рідких випадках відзначається незначна кількість еритроцитів (3–10 у полі зору). Приблизно в 1/3 хворих спостерігаються фосфатурія й лужна реакція сечі.
Набряки, артеріальна гіпертензія і зміни з боку судин очного дна не характерні для мієломної нефропатії і звичайно відсутні, навіть при розвитку ниркової недостатності. Артеріальний тиск, як правило, не підвищується і має тенденцію до зниження в міру прогресування хвороби. Набряки можливі лише в тих рідкісних випадках, коли мієломна нефропатія виявляється у вигляді амілоїдозу нирок із нефротичним синдромом.
Ниркова недостатність при мієломній хворобі зустрічається у 20–40 % хворих і розцінюється як друга за частотою причина смерті цих хворих (після інфекційних ускладнень). Розвиткові ниркової недостатності звичайно передує більш-менш тривала протеїнурія з наявністю приблизно в 1/3 хворих білка Бенс-Джонса.
Таким чином, протеїнурія і хронічна ниркова недостатність — найбільш часті і характерні прояви мієломної нефропатії. Інші ниркові синдроми й симптоми зустрічаються рідше (гостра ниркова недостатність, нефротичний синдром, синдром Фанконі).
У деяких випадках як початковий прояв мієломної нефропатії може розвитися ГНН. Причини її виникнення різні: найчастіше це результат блокування канальців білковими преципітатами або кристалізації кальцію (нефрокальциноз). Наприклад, описані випадки розвитку ГНН у хворих на мієломну хворобу безпосередньо після внутрішньовенної (екскреторної) урографії, що проводилася для уточнення причини протеїнурії неясного генезу. ГНН у таких випадках перебігає дуже тяжко й в основному має летальний кінець. Тому при підозрі на мієломну хворобу, а тим більше при вже встановленому діагнозі цього захворювання екскреторна урографія таким хворим протипоказана.
Установити діагноз мієломної нефропатії дуже складно, особливо в тих випадках, коли вона є першим або основним синдромом мієломної хвороби. Ураження нирок, що супроводжується ізольованою і стійкою протеїнурією, часто перебігає під маскою гломерулонефриту, амілоїдозу або пієлонефриту. Прижиттєва клінічна діагностика таких варіантів мієломної хвороби досить утруднена, а діагностичні помилки досягають 30–50 %. Наявність при цьому анемії і збільшення ШОЕ спочатку не знаходять належного пояснення, і лише в пізній стадії хвороби їм ретроспективно дається правильна оцінка.
Про можливість мієломної нирки необхідно думати й у тих випадках, коли протеїнурія виникає немов безпричинно (без попередньої ангіни, при відсутності в анамнезі вказівок на гострий гломерулонефрит, хронічні гнійні захворювання і т.п.) у поєднанні з анемією, високою ШОЕ, особливо якщо таке поєднання спостерігається в осіб старше 40–45 років, при відсутності набряків, артеріальної гіпертензії, гематурії і при наявності гіперпротеїнемії і гіперкальціємії. Діагноз більш переконливий, якщо згадані ознаки розвиваються на тлі кісткової патології.
Для уточнення діагнозу необхідно досліджувати сечу на білок Бенс-Джонса, провести рентгенографію кісток (черепа, ребер, здухвинних, хребців), електрофорез білків крові і сечі (з метою знайти специфічну для мієломної хвороби М-фракцію або М-градієнт) і нарешті стернальну пункцію. Що стосується пункційної біопсії нирки, то діагностична цінність цього методу є суперечливою, оскільки морфологічні зміни в нирках при мієломній хворобі доволі різноманітні, і встановити специфічні ознаки мієломної нирки вдається далеко не завжди. У той же час пункційна біопсія нирки дозволяє виключити амілоїдоз і гломерулонефрит.
Хронічна ниркова недостатність, обумовлена мієломною ниркою, на відміну від ХНН іншої етіології (зокрема, гломерулонефриту) не супроводжується раннім розвитком артеріальної гіпертензії. Рівень кальцію в крові таких хворих завжди підвищений, у тому числі й у стадії ХНН. При розвитку канальцевого ацидозу в крові підвищується рівень натрію і хлору і знижується вміст калію, тоді як із сечею знижується добова екскреція натрію, хлору, кальцію, фосфору і підвищується виділення калію.
Перебіг мієломної нефропатії, як і самої мієломної хвороби, хронічний, неухильно прогресуючий, із розвитком хронічної ниркової недостатності, що приблизно в 1/3 випадків є безпосередньою причиною смерті. При дифузних формах мієломної хвороби з тотальним ураженням кісткового мозку причиною летального результату є виражена анемія і геморагічний діатез. В інших випадках летальний кінець настає при явищах загальної кахексії або внаслідок ускладнень, пов’язаних із множинними переломами кісток, — пневмонії при переломах ребер, уросепсису у зв’язку з компресійними переломами хребців.
Середня тривалість хвороби від початку її перших клініко-лабораторних проявів становить 2–5 років і лише в окремих неускладнених випадках досягає 6–10 років.
Дотепер не існує ефективних методів і засобів лікування мієломної хвороби. Проте застосування комплексної терапії з використанням цитостатиків (циклофосфамід та ін.) у поєднанні з глюкокортикоїдами дозволяє в багатьох випадках домогтися тривалої (до 2–4 років) клінічної ремісії і, отже, збільшення тривалості життя хворого, тимчасового відновлення його фізичної активності і навіть працездатності (в осіб, не зайнятих фізичною працею).
Однак цитостатики й глюкокортикоїди можна призначати лише за відсутності ознак ниркової недостатності. У хворих на мієломну нефропатію у стадії хронічної ниркової недостатності застосування цих препаратів протипоказане. У таких випадках проводиться симптоматична терапія (як і при ХНН іншої етіології). Призначення діалізу і проведення трансплантації нирки залишається суперечливим.