
Газета «Новости медицины и фармации» Гастроэнтерология (419) 2012 (тематический номер)
Вернуться к номеру
Доброкачественные гипербилирубинемии
Авторы: И.Н. Скрыпник, д.м.н., профессор, А.С. Маслова, к.м.н. - Высшее государственное учебное учреждение Украины «Украинская медицинская стоматологическая академия», кафедра внутренней медицины № 1, г. Полтава
Версия для печати
Доброкачественные гипербилирубинемии — это группа заболеваний, которые связаны с нарушением обмена билирубина и проявляются хронической или переменной желтухой без существенных изменений структуры и функции печени и наличия признаков гемолиза и холестаза.
Этиология и патогенез
Непрямой билирубин переходит в прямой в купферовских клетках под воздействием глюкуронилтрансферазы. У больных с синдромом Жильбера этот фермент имеет сниженную активность на 25 %. Кроме повышенного гемолиза, причиной роста непрямого билирубина сыворотки крови могут быть: нарушение переноса непрямого билирубина из плазмы крови в клетки печени или нарушение связывания билирубина с глюкуроновой кислотой вследствие дефицита глюкуронилтрасферазы.
Причина повышения в сыворотке крови прямого билирубина — нарушение экскреции билирубина через мембрану гепатоцитов в желчные капилляры.
Перечисленные механизмы в тех или других случаях обусловливают развитие разных типов желтух. При доброкачественной гипербилирубинемии типа Жильбера — Мейленграхта, синдроме Криглера — Наджара появление желтухи вызвано нарушением захвата или переноса свободного билирубина из плазмы в клетки печени и связывания его с глюкуроновой кислотой. При синдроме Дабина — Джонсона и Ротора затруднение выделения уже связанного билирубина из печеночных клеток в желчные капилляры — причина его накопления в крови.
Морфологическая характеристика
1. Нормальная структура долек.
2. Воспалительные изменения отсутствуют.
3. Признаков диспротеинемии, некроза в печеночных клетках нет.
4. Накопление в печеночных клетках по ходу желчных капилляров мелкого золотистого и желто-коричневого пигмента.
5. Отсутствуют признаки развития соединительной ткани, фибротизации.
6. Иногда в пространстве Диссе незначительное накопление основного вещества, но волокнистые образования не встречаются.
Общая клиническая симптоматика
Заболевание чаще диагностируют в юношеские годы. Мужчины болеют в 10 раз чаще женщин. Более чем у 80 % больных выявляется наследственный характер заболевания.
Основные признаки при всех формах гипербилирубинемий:
— иктеричность склер (желтое окрашивание наблюдается редко) носит переменный характер;
— частичное желтушное окрашивание ладоней, подошв ног, паховых участков, носогубного треугольника;
— редко встречаются ксантелазмы и ксантомы;
— боль и тяжесть в правом подреберье (особенно в период усиления иктеричности);
— тошнота, отсутствие аппетита, отрыжка;
— запоры или поносы;
— астеновегетативные расстройства: депрессия, снижение концентрации внимания, быстрая утомляемость, слабость, обмороки, потливость, бессонница, неприятные ощущения в области сердца;
— гепатомегалия у 1/3 больных (печень выступает на 1–2 см, редко на 3–4 см из-под края реберной дуги, мягкая, безболезненная при пальпации).
Желтушное окрашивание кожи встречается не у всех больных, не сопровождается зудом, при этом часто легкая желтушность кожи остается незамеченной. Желтушность склер и кожи носит переменный характер (редко постоянная), усиливается при нервном и физическом переутомлении, у 1/3 больных — при обострении инфекции желчных путей, простудных заболеваниях, нарушении диеты, голодании, употреблении алкоголя. На коже лица могут появляться телеангиэктазии — небольшие пятна, имеющие форму сеточек или звездочек, заполненные сетью мелких и видных под кожей кровеносных сосудов. Течение заболевания хроническое, с обострениями.
У всех больных определяется гипербилирубинемия. От преобладания фракции билирубина зависит вид синдрома доброкачественной гипербилирубинемии.
Белковые осадочные пробы, протромбин не изменены, и лишь у больных с сопутствующей инфекцией в желчных путях наблюдается незначительное повышение a- и g-глобулинов. Активность АлАТ, АсАТ и щелочной фосфатазы не отличается от показателей практически здоровых лиц.
Проба с бромсульфалеином малоинформативна.
Информативно изучение поглотительной и выделительной функции печени с бенгальским розовым, меченым 131I:
— удлиняется полупериод клиренса в среднем до 28 мин против 13 мин у практически здоровых лиц;
— увеличивается время максимального поглощения — 56 мин против 25 мин в норме;
— замедляется экспрессия краски — 4,2 ч против 1,5 ч у здоровых.
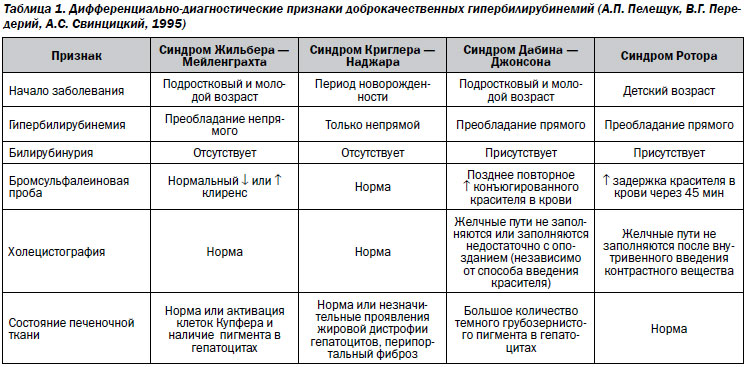
Дифференциальная диагностика
Гипербилирубинемия типа Жильбера — Мейленграхта
Характеризуется повышением концентрации билирубина крови на 30–40 % с преобладанием его непрямой фракции. У большинства больных она носит семейный характер с аутосомно-рецессивным типом наследования.
Синдром Жильбера — разновидность доброкачественной непрямой гипербилирубинемии, обусловленной наследственным дефектом промоторной зоны гена UGT1A1 (последовательность ТАТАА на 2-й паре хромосом), который кодирует уридиндифосфатглюкуронилтрансферазу (УДФГТ), определяющую метаболизм билирубина.
Гомозиготное носительство UGT1A1 составляет 5–10 %, а гетерозиготное — 40–45 %. У 38,6 % больных синдром Жильбера сочетается с эссенциальным тремором, который может указывать на наличие генетической связи между ними. Активность УДФГТ снижается при всех видах доброкачественных гипербилирубинемий. Болеют преимущественно лица молодого возраста (58–70 %); соотношение мужчин и женщин — 7 : 1.
С диагностической целью целесообразно проводить пробы с низкокалорийной диетой (400 ккал/сутки в течение 3 суток) с последующим назначением фенобарбитала (0,1 г/сутки на ночь на протяжении недели). Диагностическую значимость функциональных тестов следует оценивать по чувствительности и специфичности в зависимости от динамики уровня билирубина.
Проба с низкокалорийной диетой. Чувствительность теста определяется как процент больных, которые ответили увеличением концентрации билирубина на 21,4 мкмоль/л (в случаях гомозиготного носительства UGT1A1). Чувствительность пробы составляет 73 %, а специфичность — 100 %.
Проба с фенобарбиталомспособствует уменьшению уровня билирубина до 23,6 мкмоль/л. Проба позитивная, если уровень билирубина снижается более чем в 3 раза от его значений после пробы с низкокалорийной диетой. Чувствительность теста с фенобарбиталом — 82,9 % (достаточно высокая), специфичность — 63,3 % (низкая).
Целесообразно применение двух проб: в случаях умеренного повышения уровня билирубина на низкокалорийную диету (10–21,4 %) чувствительность при последовательном применении тестов — 78,4 %, специфичность — 96,7 %.
В настоящее время при синдроме Жильбера стало возможным определение количества ТА-повторов в промоторной области гена UGT1A1 (UDP-glycosyltransferase 1 family, polypeptide A1 gene). В норме 6 повторов в промоторной области гена UGT1A1. Увеличение количества повторов в этой области свидетельствует о снижении функциональной активности UGT1A1.
Клиническая классификация синдрома Жильбера (Л.Ю. Ильченко и соавт., 2006)
І. В зависимости от варианта клинического течения:
— диспептический;
— астеновегетативный;
— желтушный;
— латентный.
ІІ. По генотипу (полиморфизму промоторного участка гена UGT1A1):
— гетерозиготное носительство;
— гомозиготное носительство.
ІІІ. По состоянию детоксикационной функции печени:
— с сохраненной функцией;
— со сниженной функцией.
ІV. В зависимости от ассоциации с заболеваниями:
— с эссенциальным тремором;
— без эссенциального тремора.
Синдром Дабина — Джонсона
Характеризуется нарушением экскреции пигмента из гепатоцитов и приводит к дизрегуляции обмена билирубина. В сыворотке крови находится только прямой билирубин или преобладает его фракция. Кроме этого, нарушено выделение бромсульфалеина и рентгеноконтрастных препаратов, отсутствует тень желчного пузыря при холецистографии.
Клиническая симптоматика более выражена, чем при других формах гипербилирубинемии. Желтуха постоянная, сопровождается незначительным зудом кожи. Часто возникают диспептические расстройства, общее самочувствие всегда нарушено.
Диагностические критерии:
— накопление пигмента в гепатоцитах в виде аморфных гранул диаметром от 0,5 до 4 мкм, которые содержат липофусцин;
— повышение концентрации бромсульфалеина в крови через 2 часа после начала наблюдения;
— удлинение полупериода экскреции бенгальського розового, меченого 131I (до 7 часов).
Синдром Ротора
По клиническим признакам не отличается от синдрома Дабина — Джонсона, но характеризуется отсутствием накопления липофусцина в клетках печени, хотя многочисленные гепатоциты содержат мелкие и средние по размерам капли жира.
Диагностические критерии:
— значительная длительность желтухи;
— преобладание преимущественно прямой фракции билирубина;
— отсутствие пигмента при гистологическом исследовании;
— достаточное заполнение желчного пузыря при контрастной рентгенографии;
— умеренное снижение поглощения и выделения желчи печенью при исследовании с бенгальским розовым.
Синдром Криглера — Наджара
Связан с полной или почти полной неспособностью печени конъюгировать билирубин вследствие дефицита глюкуронилтрансферазы. Существуют генетически гетерогенные формы заболевания.
I форма — передается по аутосомно-рецессивному типу; характерна интенсивная желтуха за счет повышения уровня непрямого билирубина крови в 15–50 раз выше нормы. Гипербилирубинемия сохраняется на протяжении всей жизни, она резистентна к фенобарбиталу.
II форма — наследуется по аутосомно-доминантному типу и сопровождается более выраженной желтухой с 5–20-кратным повышением уровня непрямого билирубина. Желчь окрашена, в кале — значительное количество уробилиногена. Фенобарбитал уменьшает концентрацию билирубина сыворотки, его высокий уровень обусловливает неврологическую симптоматику.
Прогноз
Имеет благоприятный характер. Больные живут до старости, особенно те, которым проводится профилактическое лечение. Обострение заболеваний провоцируют преимущественно нервно-психические перенапряжения, а также простудные заболевания, обострения инфекций желчных путей.
Лечение
В специфическом лечении больные с доброкачественными гипербилирубинемиями не нуждаются.
Важно соблюдение режима труда и отдыха, питания. Режим больных должен быть облегченным. Противопоказан труд, связанный со значительным психическим и физическим напряжением. Следует избегать ограничения жидкости, голодания и гиперинсоляций. Резко ограничивается прием медикаментов, исключается алкоголь. Следует помнить, что синдром Жильбера не является поводом для отказа от прививок.
В фазу ремиссии можно назначить диету № 15 по Певзнеру с исключением мяса жирных сортов и консервов, острых блюд и пряностей. При наличии других заболеваний желчевыводящих путей показана диета № 5.
При синдроме Жильбера рекомендуется назначение урсодезоксиксихолевой кислоты (урсофалька) в дозе 15–25 мг/кг/сутки, s-адеметионина (гептрала) в дозе 800–1200 мг/сутки, гепазила композитума по 1 ампуле для питья в сутки в утренние часы. Показана витаминотерапия (витамины группы В). Больным с синдромом Жильбера необходимо назначать любые фармакологические препараты строго по показаниям, учитывая необходимость максимального щажения ксенобиотической функции печени.
В фазу обострения синдрома Жильбера и синдрома Криглера — Наджара II типа рекомендуется назначение фенобарбитала или зиксорина в дозе 30–180 мг/сутки на протяжении 2–4 недель. Фенобарбитал и зиксорин индуцируют синтез микросомальных ферментов и особенно ферментов, которые осуществляют конъюгацию, повышают активность глюкуронилтрансферазы. Глюкокортикоиды ухудшают состояние больных.
Больным с синдромом Криглера — Наджара I типа рекомендуется терапия с использованием ламп дневного света, солнечного света, введение раствора альбуминов, обменные гемотрансфузии.
Лечение больных с синдромом Дабина — Джонсона и Ротора не разработано.
Специальное курортное лечение доброкачественных гипербилирубинемий не показано. Следует учитывать, что тепловые и электрические физпроцедуры на область печени вредны.
1. Гастроентерологія / Харченко Н.В., Бабак О.Я. та ін. — К.: Друкар, 2007. — 720 с.
2. Гастроэнтерология и гепатология: диагностика и лечение: Рук-во для врачей / Под ред. А.В. Калинина, А.И. Хазанова. — М.: Миклош, 2007. — 602 с.
3. Дегтярева И.И. Клиническая гастроэнтерология. — М.: Междун. информ. агентство, 2004. — 616 с.
4. Ильченко Л.Ю., Дроздов В.Н., Шулятьев И.С. Синдром Жильбера: клинико-генетическое исследование // Терапевт. арх. — 2006. — Т. 78, № 2. — С. 48-52.
5. Подымова С.Д.. Болезни печени. Руководство для врачей. — 4-е изд. — М.: Медицина, 2005. — 768 с.
6. Скрипник І.М., Мельник Т.В., Потяженко М.М. Клінічна гепатологія. — Полтава: Дивосвіт, 2007. — 424 с.
7. Шерлок Ш., Дули Дж. Заболевания печени и желчных путей: Практич. рук.: Пер. с англ. / Под ред. З.Г. Апросиной, Н.А. Мухина. — М.: ГЭОТАР-медицина, 2002. — 864 с.