
Газета «Новости медицины и фармации» 18 (472) 2013
Вернуться к номеру
Второй механизм действия симвастатина: подавление воспаления в атеросклеротической бляшке
Авторы: Савустьяненко А.В. - доц. кафедры фармакологии
Донецкого национального медицинского университета им. М.Горького
Рубрики: Кардиология
Разделы: Клинические исследования
Версия для печати
 Конец XX — начало XXI века можно, бесспорно, считать временем распознавания воспаления как наиболее универсального патологического процесса. В многочисленных исследованиях было показано, что воспаление сопровождает большинство из известных нам заболеваний, включая онкологию и психические расстройства. С учетом этого и поиск лекарственных средств был расширен до тех препаратов, которые, наряду со специфическим для данной болезни эффектом, обладали бы способностью подавлять и сопровождающее ее воспаление.
Конец XX — начало XXI века можно, бесспорно, считать временем распознавания воспаления как наиболее универсального патологического процесса. В многочисленных исследованиях было показано, что воспаление сопровождает большинство из известных нам заболеваний, включая онкологию и психические расстройства. С учетом этого и поиск лекарственных средств был расширен до тех препаратов, которые, наряду со специфическим для данной болезни эффектом, обладали бы способностью подавлять и сопровождающее ее воспаление.
Учитывая высокую распространенность атеросклероза и связанных с ним сердечнососудистых осложнений, мы бы хотели представить вниманию читателей обзор работы японских исследователей [Tahara N. и соавт.], посвященной оценке воспаления при атеросклерозе и противовоспалительного действия симвастатина.
Что мы знаем о воспалении при атеросклерозе?
Еще совсем недавно предложенный механизм развития атеросклероза выглядел довольно простым. Считалось, что, с одной стороны, он представляет собой процесс пассивного накопления холестерина в местах поврежденного эндотелия. С другой, агрегация тромбоцитов к тем же самым местам приводит к высвобождению тромбоцитарного фактора роста, который стимулирует размножение гладкомышечных клеток. Вместе накопление липидов и пролиферация гладкомышечных клеток способствуют формированию и росту атеросклеротических бляшек.
Однако в исследованиях, выполненных в последние десятилетия, было показано, что в формировании атеросклероза принимают также участие клетки неспецифического (моноциты/макрофаги, базофилы, тромбоциты) и специфического (Тклетки) иммунитета.
Моноциты с помощью специальных молекул адгезии проникают через эндотелий к местам атерогенеза и превращаются в макрофаги. Известно, что популяция моноцитов в организме неоднородна: имеются провоспалительные и противовоспалительные моноциты. Было убедительно показано, что при атеросклерозе задействуется именно провоспалительный пул (Ly6C). Превращаясь в макрофаги, эти клетки секретируют большое число медиаторов (например, цитокинов), запускающих и поддерживающих воспаление (рис. 1).
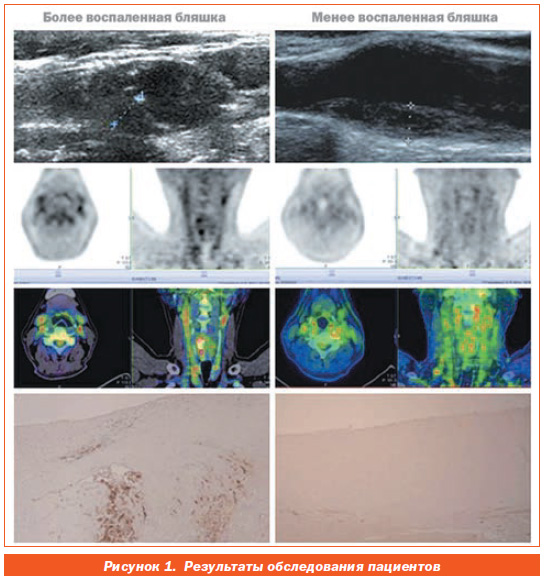
На рис. 1 представлены результаты обследования двух пациентов (левая и правая колонки). Видно, что у них одинаковый размер стенотических бляшек в сонных артериях (пунктирные линии на верхних рисунках). Однако выраженность воспаления в бляшке у одного пациента (слева) больше, чем у другого (справа). Об этом свидетельствуют более интенсивный сигнал PET (позитронноэмиссионная томография; цветной рисунок) и более выраженная инфильтрация макрофагов (нижний рисунок) у пациента, представленного слева.
Базофилы известны своим участием в аллергических процессах. Однако было показано, что, секретируя малые вазоактивные молекулы, такие как гистамин и лейкотриены, ряд сериновых протеаз и гепарин, они также участвуют и в формировании атеросклероза.
Активированные тромбоциты, помимо участия в тромбообразовании, могут высвобождать преформированные (синтезированные на этапе созревания тромбоцитов) провоспалительные цитокины и миелоидсвязанный белок 8/14 (MRP8/14). Эти медиаторы способствуют поддержанию воспаления. При изучении атеросклероза, возможно, впервые была показана взаимосвязь между тромбозом и воспалением.
Одна из популяций мононуклеарных фагоцитов — дендритные клетки — также проникает внутрь формирующихся бляшек и предоставляет антигены для Тклеток. В результате последние начинают высвобождать провоспалительные цитокины, усиливая воспаление, а также проявлять цитотоксическое (киллерное) действие.
Результатом вовлечения всех вышеописанных клеток является усиление воспаления при атеросклерозе, которое опосредует различные этапы формирования атеросклеротических бляшек, вплоть до их разрыва. Соответственно, подавление воспаления могло бы уменьшить прогрессирование атеросклероза и способствовать стабилизации бляшек.
В клинике хорошим биомаркером выраженности воспаления при атеросклерозе считается Среактивный белок, измеренный высокочувствительным методом (CRPhs). Если у индивидуума нет острых инфекций и воспалительных заболеваний (например, ревматоидного артрита), уровень CRPhs остается стабильным в течение длительного периода времени, и его межгодовая и междекадная вариабельность сравнима с таковой у холестерина. Важно и то, что для него не характерна вариация в течение суток.
Дизайн японского исследования
Известно, что статины — одна из наиболее популярных лекарственных групп для лечения атеросклероза — способны снижать уровень атерогенных и повышать уровень антиатерогенных липопротеидов в плазме крови, препятствуя развитию атеросклероза. В дополнение к этому было показано, что статины способны снижать выраженность показателей системного воспаления (CRPhs). Однако не было выполнено исследований с визуализацией, в которых была бы продемонстрирована способность статинов подавлять воспаление непосредственно в атеросклеротической бляшке.
Для того чтобы ответить на вопрос, проявляют ли статины подобный эффект, японская группа исследователей выполнила проспективное рандомизированное контролируемое 3месячное исследование. В него в общей сложности было включено 43 пациента, из которых 22 находились только на специальной диете, а 21 в дополнение к такой же диете получали симвастатин. В группе с симвастатином стартовая доза препарата составляла 5–10 мг/день. Если спустя 1 месяц лечения уровень холестерина липопротеидов низкой плотности (ЛПНП) превышал 130 мг/дл (3,4 ммоль/л), то дозу препарата увеличивали до 20 мг/день — максимальная доза симвастатина, одобренная в Японии. В результате 2 пациента получали 5 мг/день, 14 пациентов — 10 мг/день, 5 пациентов — 20 мг/день препарата.
В качестве метода визуализации была выбрана позитронноэмиссионная томография, в качестве зонда — 18Fфтордезоксиглюкоза (18FDG). Идея использования этого зонда связана с тем, что он является аналогом естественного метаболита — глюкозы и активно захватывается клетками, в которых интенсивно протекает обмен веществ. К ним относят, например, опухолевые клетки, клетки мозга, а также воспалительные клетки. В более ранних исследованиях было показано, что 18FDG захватывается инфильтрирующими воспалительными клетками (макрофаги, лимфоциты) и субэндотелиально пролиферирующими макрофагами и гладкомышечными клетками в местах атеросклеротического повреждения.
Поскольку PET имеет ограниченное пространственное разрешение, выполняли его совместную регистрацию вместе с компьютерной томографией (КT), что позволяло очень точно локализовать место захвата 18FDG. Например, совместная регистрация PET и КT при таком системном заболевании, как синдром дуги аорты, позволяла выявлять активное сосудистое воспаление с чувствительностью 90,9 %, специфичностью 88,8 %.
Для того чтобы количественно оценить захват 18FDG воспалительными клетками атеросклеротических бляшек, рассчитывали стандартизированное значение захвата (SUV) (при этом ориентировались на максимальную пиксельную активность PET внутри интересующей области). Величину SUV оценивали в каждой из отдельных бляшек каждого из пациентов, а затем усредняли, что позволяло сравнить значение этого показателя в исходных условиях и спустя 3 месяца лечения.
Помимо исследования с визуализацией, была выполнена также оценка стандартных биохимических показателей, позволявших судить о влиянии симвастатина на динамику атеросклероза: холестерин ЛПНП, холестерин липопротеидов высокой плотности (ЛПВП), уровень CRPhs и т.д.
Результаты исследования
Визуализация с помощью PET
В исходных условиях было обнаружено 117 и 123 воспалительные бляшки в сонных артериях и аорте пациентов, получавших симвастатин, и пациентов, получавших только диету, соответственно. Между обеими группами пациентов не было обнаружено достоверных различий по SUV.
Спустя 3 месяца лечения симвастатин ослаблял захват 18FDG в атеросклеротических бляшках, в то время как сама по себе диета не приводила к подобному результату (рис. 2). Достоверное снижение SUV наблюдалось только в группе с симвастатином (p < 0,01).
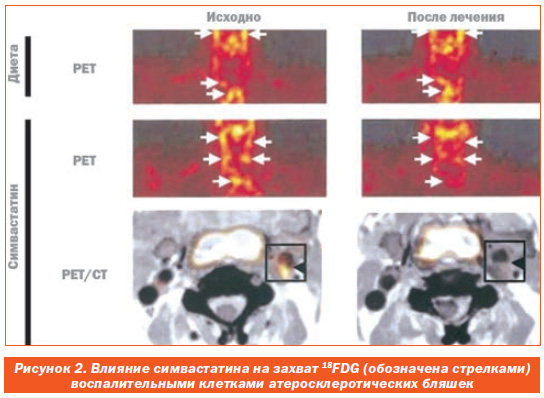
Таким образом, поскольку визуальная оценка захвата 18FDG и количественная оценка показателя SUV отражают выраженность воспаления, и симвастатин снижал оба этих параметра, то авторами исследования был сделан вывод о том, что симвастатин обладает противовоспалительным действием и подавляет протекание воспалительной реакции внутри атеросклеротических бляшек.
На рис. 2 представлены репрезентативные PETизображения в исходных условиях и спустя 3 месяца лечения только диетой или симвастатином вместе с диетой. Диета сама по себе не влияла на захват 18FDG в дуге аорты и сонных артериях. Захват 18FDG был ослаблен на фоне лечения симвастатином. Совместная регистрация PET и КT отчетливо показывает, что захват бляшкой 18FDG прекращался спустя 3 месяца лечения симвастатином.
Влияние на биохимические показатели
В исходных условиях профиль липидов был одинаков в обеих группах. Спустя 3 месяца лечения симвастатин снижал уровень холестерина ЛПНП на 30 % (p < 0,01) и увеличивал содержание холестерина ЛПВП на 15 % (p < 0,01). В группе пациентов, получавших только диету, достоверных изменений отмечено не было.
Ни в одной из групп не было обнаружено достоверных изменений CRPhs, что, повидимому, объясняется малым размером групп.
Дополнительно было выяснено, что снижение SUV в группе с симвастатином хорошо коррелировало с увеличением содержания холестерина ЛПВП (p < 0,01), в то время как подобная корреляция не была обнаружена для холестерина ЛПНП. В группе, получавшей только диету, не было обнаружено ни одной из подобных корреляций.
Таким образом, в исследовании был зарегистрирован ожидаемый эффект симвастатина на основные биохимические показатели липидного обмена. Авторы отмечают, что отсутствие корреляции между снижением SUV и снижением холестерина ЛПНП указывает на то, что противовоспалительный эффект симвастатина не зависит от снижения содержания атерогенной фракции липидов в плазме крови. Возможно, этот эффект опосредуется повышением холестерина ЛПВП, а возможно — какимито другими механизмами.
Выводы
1. Результаты новых исследований свидетельствуют о том, что атеросклероз — это не только накопление атерогенных липопротеидов на фоне пролиферации гладкомышечных клеток. Был продемонстрирован воспалительный характер данного заболевания с вовлечением в процесс клеток неспецифического (моноциты/макрофаги, базофилы, тромбоциты) и специфического (Тклетки) иммунитета.
2. В работе японской группы исследователей было убедительно показано, что симвастатин в дополнение к классическому эффекту — снижению уровня атерогенных и повышению уровня антиатерогенных липопротеидов — подавляет течение воспалительных процессов в бляшках при атеросклерозе.
3. Противовоспалительный эффект симвастатина был независим от снижения уровня холестерина ЛПНП, что позволяет рассматривать этот эффект как дополнительный, отдельный.
1. Simvastatin attenuates plaque inflammation: evaluation by fluorodeoxyglucose positron emission tomography / Tahara N., Kai H., Ishibashi M. et al. // J. Am. Coll. Cardiol. — 2006. — V. 48, № 9. — P. 18251831.
2. Libby P., Ridker P.M., Hansson G.K. Inflammation in atherosclerosis: from pathophysiology to practice // J. Am. Coll. Cardiol. — 2009. — V. 54, № 23. — P. 21292138.
3. Hwang B.H., Kim M.H., Chang K. Molecular imaging of highrisk atherosclerotic plaques: is it clinically translatable? // Korean Circ. J. — 2011. — V. 41, № 9. — P. 497502.