
Журнал «Медицина неотложных состояний» 3 (58) 2014
Вернуться к номеру
Клинико-прогностическое значение полиморфизма гена эндотелиальной NO-синтетазы у больных с острыми коронарными синдромами
Авторы: Пархоменко А.Н., Лутай Я.М., Иркин О.И., Кожухов С.Н., Скаржевский А.А. - ННЦ «Институт кардиологии имени академика Н.Д. Стражеско» НАМН Украины, г. Киев; Досенко В.Е., Мойбенко А.А. - Институт физиологии им. А.А. Богомольца НАН Украины, г. Киев
Рубрики: Медицина неотложных состояний, Кардиология
Разделы: Клинические исследования
Версия для печати
Оксид азота (NO) производится эндотелиальной синтетазой оксида азота (eNOS) и играет важную роль в поддержании сосудистого гомеостаза. Были определены различные полиморфизмы гена eNOS. -786С полиморфизм промотора гена модулирует экспрессию гена eNOS, а значит, синтез NO.
Обследованы 325 больных с острым коронарным синдромом (ОКС) и 83 здоровых пациента (контрольная группа). Полиморфизм eNOS был проанализирован с помощью методов полимеразной цепной реакции и полиморфизма длин рестрикционных фрагментов. У украинцев генотип C/C промотора гена eNOS встречается чаще, чем у японцев, и реже, чем у жителей Западной Европы, Северной Америки и Австралии. Значительная разница в распределении генотипа наблюдалась между исследуемыми пациентами и группой контроля. Патологический C/C генотип значительно чаще определяется у больных с ОКС, особенно у мужчин моложе 55 лет.
Наличие -786CC генотипа было связано с увеличением артериальной жесткости и снижением поток-опосредованного расширения плечевой артерии. Реперфузионная терапия была эффективнее у пациентов с -786TT вариантом промотора гена eNOS. Наличие -786TT генотипа обусловливало меньшее количество осложнений в период госпитализации.
Оксид азоту (NO) виробляється ендотеліальною синтетазою оксиду азоту (eNOS) і відіграє важливу роль у підтримці судинного гомеостазу. Було визначено різні поліморфізми гена eNOS. -786С поліморфізм промотора гена модулює експресію гена eNOS і відповідно — синтез NO.
Обстежені 325 хворих із гострим коронарним синдромом (ГКС) і 83 здорових пацієнти (контрольна група). Поліморфізм eNOS проаналізовано за допомогою методів полімеразної ланцюгової реакції й поліморфізму довжин рестрикційних фрагментів. В українців генотип C/C промотора гена eNOS зустрічається частіше, ніж у японців, і рідше, ніж у мешканців Західної Європи, Північної Америки й Австралії. Значна різниця в розподілі генотипу спостерігалася між досліджуваними пацієнтами й групою контролю. Патологічний C/C генотип значно частіше визначається у хворих із ГКС, особливо в чоловіків молодше 55 років.
Наявність -786CC генотипу була пов’язана із збільшенням артеріальної жорсткості й зниженням потік-опосередкованого розширення плечової артерії. Реперфузійна терапія була ефективнішою в пацієнтів із -786TT варіантом промотора гена eNOS. Наявність -786TT генотипу обумовлювала меншу кількість ускладнень у період госпіталізації.
Nitric oxide (NO) is produced by endothelial nitric oxide synthetase (eNOS) and plays important role in normal vascular homeostasis. In the eNOS gene various polymorphisms have been identified. The -786T>C polymorphism in the gene promoter has been demonstrated to modulate the eNOS gene expression and so the synthesis of NO.
325 patients with ACS and 83 healthy controls were investigated. The eNOS polymorphism has been analyzed by polymerase chain reaction and restriction fragment length polymorphism. In Ukrainians the C/C genotype in the eNOS gene promoter was more common than in Japanese and less common than in west Europeans, north Americans and Australians. A significant difference in genotype distribution was observed between patients and controls. Pathological C/C genotype was significantly more often determined in ACS patients, especially in men younger than 55 years old.
-786CC genotype was associated with increase in arterial stiffness and lower flow-mediated brachial artery dilatation. -786TT variant of the eNOS gene promoter in investigated patients was associated with better efficacy of reperfusion therapy. Patients with -786TT genotype had fewer complications during the period of hospitalization.
полиморфизм гена, эндотелиальная NO-синтетаза, эндотелиальная дисфункция, скорость распространения пульсовой волны, острый коронарный синдром, тромболитическая терапия.
поліморфізм гена, ендотеліальна NO-синтетаза, ендотеліальна дисфункція, швидкість поширення пульсової хвилі, гострий коронарний синдром, тромболітична терапія.
поліморфізм гена, ендотеліальна NO-синтетаза, ендотеліальна дисфункція, швидкість поширення пульсової хвилі, гострий коронарний синдром, тромболітична терапія.
Статья опубликована на с. 45-54
Эндотелий сосудистой стенки играет важную роль в поддержании сосудистого гомеостаза. Посредством синтеза эндотелийзависимых факторов релаксации и констрикции происходит контроль тонуса сосудистой стенки, тромбообразования, клеточной пролиферации и атерогенеза [3, 4, 25]. Ключевое значение в реализации защитных функций эндотелия имеет оксид азота (NO), который синтезируется эндотелиальной NO-синтетазой [2, 17, 32]. NO-синтетаза принадлежит к семейству оксидоредуктаз. В настоящее время описаны три изоформы NO-синтетаз: нейрональная изоформа (nNOS, NOS1), макрофагальная, или индуцибельная, изоформа (iNOS, NOS2) и эндотелиальная изоформа (eNOS, NOS3) [9, 28, 30, 31]. Эти ферменты только на 50–60 % гомологичны по своему аминокислотному составу и кодируются разными генами, находящимися в разных хромосомах [29, 30, 41]. Эндотелиальная и нейрональная изоформы являются конституциональными разновидностями фермента, а индуцибельная NO-синтетаза экспрессируется в основном при воспалении или инфекционном процессе [28].
Ген, кодирующий eNOS, находится в хромосоме 7 (q35–36) и состоит из 26 экзонов [23, 30]. Промотор гена eNOS содержит несколько доменов, то есть может регулироваться рядом факторов транскрипции [27, 30, 31, 49]. В 1995 году Hingorani et al. было высказано предположение о наличии полиморфизма гена, кодирующего эндотелиальную NO-синтетазу [18, 19]. Наиболее изученными являются 4a/b полиморфизм четвертого интрона, G894T (Glu298Asp) полиморфизм седьмого экзона и Т-786С полиморфизм промотора гена эндотелиальной NO-синтетазы [10, 19, 35].
Наша работа посвящена изучению распространенности полиморфизма промотора Т-786С гена eNOS и его патогенетического значения у больных с острым коронарным синдромом (ОКС).
Материалы и методы
В исследование включались пациенты, поступившие в отделение реанимации и интенсивной терапии ННЦ «Институт кардиологии им. Н.Д. Стражеско» с диагнозом НС или ОИМ. Обследовано 325 пациентов с острыми коронарными синдромами (113 пациентов без элевации сегмента ST на ЭКГ и 212 пациентов с элевацией ST). Диагнозы острого ИМ и НС устанавливали на основании данных клинических, электрокардиографических и биохимических обследований согласно существующим рекомендациям [7, 8]. В исследование не включались больные с хронической сердечной недостаточностью IIБ — III ст., истинным кардиогенным шоком, тяжелой формой сахарного диабета, выраженной почечной и печеночной недостаточностью, бронхиальной астмой, нарушениями в системе гемостаза, острым нарушением мозгового кровообращения, травмой или большим хирургическим вмешательством, острыми воспалительными процессами или обострением хронических, онкологическими и системными заболеваниями. Контрольную группу составили 83 практически здоровых донора, у которых отсутствие сердечно-сосудистой патологии подтверждали путем сбора анамнестических данных, снятия электрокардиограммы и измерения артериального давления. Контрольная группа и группа больных не отличались по возрасту и соотношению полов, P > 0,05 по x2--критерию.
Для генотипирования венозную кровь набирали в стерильных условиях в моноветы объемом 2,7 мл с калиевой солью этилендиаминтетрауксусной кислоты в качестве антикоагулянта (Sarstedt, Германия), замораживали и сохраняли при температуре –20 °С. ДНК выделяли из цельной крови с использованием наборов изоген (Россия). Методом полимеразной цепной реакции с последующим анализом длины рестрикционных фрагментов определяли Т-786С полиморфизм промотора по G. Ghilardi et al. с модификациями [13]. Для этого амплифицировали участок промотора указанного гена с помощью пары специфических праймеров: прямой (sense) — 5`-CAC CTG CAT TCT GGG AAC TGTA-3` и обратный (antisense) — 5`-GCC GCA GTA GCA GAG AGAC-3`. Праймеры были синтезированы фирмой «Синтол» (Россия). Для амплификации брали 1 мкл ДНК (концентрация от 3 до 4 нг/мкл) и добавляли к смеси, содержащей 5 мкл пятикратного РСR-буфера, 1,5 мМ сульфата магния, 200 мкМ смеcи четырех нуклеотидтрифосфатов, по 20 рМ каждого из праймеров и 0,5 EД Taq-полимеразы (амплисенс, Россия), объем доводили до 25 мкл деионизированной водой. PCR проводили в термоциклере Applied Biosystems 2700 (PerkinElmer, США). Амплификация фрагмента промотора состояла из 35 циклов: денатурация — 94 °С (1 мин), отжиг праймеров — 63 °С (50 c) и элонгация — 74 °С (1 мин). В дальнейшем 6 мкл продукта амплификации фрагмента промотора инкубировали при 37 °С в течение 18 часов с 5 ЕД рестриктазы PdiI («Ферментас», Латвия) в буфере /// Y+/Tango следующего состава: 33 мМ трисацетата (рН 7,9), 10 мМ ацетата магния, 66 мМ ацетата калия, 0,1 мг/мл альбумина. Наличие в –786 положении промотора тимидина препятствует рестрикции, а при замене на цитозин PdiI расщепляет амплифицированный участок промотора (размер 125 пар оснований) на два фрагмента — 95 и 30 пар оснований. Амплификаты фрагмента промотора после рестрикции разделяли в 2,5% агарозном геле, содержащем бромистый этидий. Визуализация ДНК после горизонтального электрофореза (160 V на протяжении 40 мин) проводилась с помощью трансиллюминатора («Биоком», Россия).
Тест с реактивной гиперемией проводился на аппарате для ультразвуковой диагностики SonoAce PICO (компания Medison) при помощи сосудистого датчика HL5-9ED (7,5 МГц/40 мм). Пациенту за сутки до проведения пробы отменялись нитропрепараты (исключая короткодействующие нитраты для купирования приступов стенокардии). Проба проводилась утром натощак, после 10-минутного отдыха в положении лежа. После измерения исходного диаметра плечевой артерии (среднее значение 4 измерений с расстоянием 1 см между точками) пациенту на плечо выше участка измерения накладывалась манжета от манометра и создавалось давление в 200 мм рт.ст., которое удерживалось в течение 5 минут. Через 80 секунд после декомпрессии проводилось повторное измерение диаметра артерии и рассчитывался показатель эндотелийзависимой вазодилатации как процент прироста диаметра артерии от исходного размера [12]. Проба с реактивной гиперемией проведена у 63 больных.
Жесткость сосудистой стенки оценивалась путем измерения скорости распространения пульсовой волны (СРПВ) на аппарате Complior® SP (ARTECH medical, Франция) [6].
Клиническое течение ОИМ оценивали по числу осложнений госпитального периода заболевания. ОЛЖН регистрировали согласно классификации T. Killip. Hарушения сердечного ритма регистрировали путем суточного мониторирования ЭКГ по методу Холтера с использованием кардиорегистратора «Ритм» (НТО «Бета», Украина) и прикроватных кардиомониторов Sirecust 342R фирмы Siemens, Германия. Пациентам проводилось серийное определение активности сывороточной КФК и МВ-КФК в периферической венозной крови в первые трое суток заболевания. При развитии рецидива или затяжном течении ОИМ длительность исследования КФК и ее МВ-фракции увеличивалась. О наступлении реканализации ИОКА свидетельствовало достижение пика активности МВ-КФК в первые 12 часов от начала заболевания [20].
Статистический анализ результатов проводили с использованием электронных таблиц Microsoft® Excel 2000 и статистических программ SPSS (версия 12, США). При этом достоверность отличий определяли по x2-критерию. Значение p < 0,05 считали достоверным.
Результаты
Проведенное генотипирование Т-786С промотора гена эндотелиальной NO-синтетазы выявило распределение генотипов –786ТТ, –786ТС и –786СС как 42,5; 41,2 и 16,3 % у больных с ОКС и как 48,2; 45,8 и 6,0 % в группе контроля (табл. 1).
/47/47.jpg)
Наблюдаемое распределение частот различных генотипов в группе обследованных больных и группе контроля соответствовало равновесию Харди — Вейнберга.
Таким образом, полиморфизм промотора гена эндотелиальной NO-синтетазы в позиции 786 определялся у 57,5 % пациентов с ОКС, а 16,3 % больных были гомозиготами с наличием патологического –786СС генотипа. При отсутствии отличий между группами по частоте встречаемости генотипов ТТ и ТС вероятность определения СС генотипа промотора была достоверно выше у больных с ОКС, чем в группе контроля (16,3 % у больных с ОКС против 6,0 % в группе контроля, р < 0,01). Среди больных c ОКС без элевации сегмента ST примерно в 4 раза чаще, чем в группе контроля (22,2 % против 6,0 %, р < 0,01), и в 2 раза чаще, чем среди больных с элевацией сегмента ST (22,2 % против 13,2 %, р < 0,05), выявляли гомозигот с патологическим СС генотипом промотора гена eNOS. Полученные данные позволяют предположить патогенетическое значение данного полиморфизма в развитии острых форм ишемической болезни сердца, особенно у больных без стойкой элевации сегмента ST на электрокардиограмме.
Дальнейший анализ этой категории больных выявил тенденцию к увеличению частоты выявления носителей аллели С в младших возрастных группах (рис. 1).
/47/47_2.jpg)
Эта зависимость была более характерна для мужчин (табл. 2).
/48/48.jpg)
Так, у мужчин с ОКС без элевации ST в возрасте до 55 лет (с преждевременным развитием ИБС) достоверно чаще выявляли патологическую аллель С (генотипы СС + ТС), чем у больных более пожилого возраста (77,8 и 51,9 % соответственно, Р < 0,01). Именно для этой категории больных характерно участие вазоспастических реакций в патогенезе острых форм ИБС, развитие которых, по данным литературы, отмечалось при мутации промотора гена eNOS [7, 8, 37].
Различий в частоте выявления полиморфизма промотора гена eNOS в других клинико-анамнестических подгруппах пациентов с ОКС не отмечено (Р > 0,05).
Согласно данным метаанализа, основанного на результатах 7 исследований, были отмечены достоверные отличия по частоте встречаемости гомозигот СС промотора гена eNOS среди здорового населения в различных этнических группах (1,1 % — для азиатского, 15,36 % — для неазиатского населения, Р < 0,0001) [10]. Данные литературы относительно роли этого полиморфизма в развитии ИБС и ее острых проявлений противоречивы [5, 10, 11, 14, 36–38, 43, 48].
Популяционное исследование, проведенное в 9 различных регионах Великобритании, показало, что соотношение нормальных гомозигот, гетерозигот и патологических гомозигот при анализе полиморфизма Т–786С промотора гена eNOS составило соответственно 37,7; 47,8 и 14,5 %. Не было отмечено влияния данного полиморфизма на развитие ИБС [24]. Распространенность различных вариантов этого полиморфизма у итальянцев соответствует таковому в Великобритании [11, 14]. При этом риск развития ИБС был достоверно выше у пациентов с –786СС, чем с –786ТТ генотипом (OR = 2,5 (1,3–4,8), Р < 0,01). Генотип СС был также независимым фактором риска развития коронарного атеросклероза [11]. Сам факт наличия патологической аллели С являлся фактором риска ИБС (OR = 1,7 (1,1–2,8), Р = 0,02 при сравнении с генотипом ТТ), а среди больных с ИБС носители этой аллели имели более выраженное поражение венечных артерий по данным коронароангиографии [11]. В исследовании G. Ghilardi и соавторов у больных, прооперированных по поводу стеноза внутренней сонной артерии, генотип СС обнаруживали в два раза чаще, чем в группе контроля (26 и 13 % соответственно, Р = 0,018), а у больных со стенозом сонной артерии генотип СС был более распространен в группе с изъязвленной атеросклеротической бляшкой (44 и 17 % соответственно, Р = 0,003) [14]. Встречаемость патологических гомозигот СС у испанцев моложе 60 лет составляет 14,3 %, в то же время среди курящих мужчин моложе 50 лет с ИБС этот генотип обнаруживали в 21,8 % случаев (Р = 0,039) [5]. Это послужило основанием предположить, что генотип СС промотора гена еNOS определяет повышенный риск преждевременного развития атеросклероза в условиях воздействия неблагоприятных факторов внешней среды (курение). Соотношение различных вариантов генотипа (ТТ, ТС и СС) промотора гена eNOS у канадцев среднего возраста без ИБС в анамнезе было близким к таковому у европейцев и распределялось как 38,9; 46,1 и 15,0 % [21]. У лиц с генотипом СС отмечали достоверно более высокие уровни систолического артериального давления, а также чаще диагностировали артериальную гипертензию. В японской популяции встречаемость аллели С, по данным исследования Suita, достаточно низкая (20,2 % населения), а встречаемость патологических гомозигот (СС) составляет около 1 % всего населения [22]. У больных с ИМ распространенность различных вариантов промотора не отличалась от таковой в общей популяции, что позволило сделать вывод об отсутствии роли этого полиморфизма в патогенезе острого ИМ у японцев [43]. Однако в исследованиях M. Nakayama и соавторов мутация Т-786С ассоциировалась с коронарным спазмом и чаще выявлялась у больных с ИМ, особенно без органического стеноза венечных артерий по данным ангиографии [36, 37].
Таким образом, патологический генотип СС промотора гена еNOS встречается у 6,0 % здоровых доноров в Украине, что достоверно больше, чем в японской популяции, и меньше, чем у западных европейцев (итальянцев, англичан, испанцев, французов), белых североамериканцев и австралийцев (табл. 3).
Для уточнения механизмов реализации мутантного генотипа в патогенезе ИБС, и ОКС в частности, мы изучили степень жесткости артериальной сосудистой стенки и выраженность эндотелиальной дисфункции у части обследованных больных.
Степень эндотелиальной дисфункции у обследованных больных с ОКС оценивалась по результатам манжеточной пробы с реактивной гиперемией. Обследованы 63 больных. Распределение –786ТТ, –786ТС и –786СС генотипов промотора гена eNOS соответствовало 46,0; 36,5 и 17,5 %. По основным вариантам генотипа и анамнестическим данным данная выборка больных не отличалась от общей группы.
Выявлена достоверно более высокая степень прироста диаметра плечевой артерии у больных с –786ТТ генотипом (8,03 ± 0,71 % у больных с генотипом ТТ по сравнению с 5,55 ± 0,92 % у больных с генотипом ТС (р < 0,05) и 5,30 ± 1,21 % у больных с генотипом СС (p < 0,05)). Таким образом, полиморфизм гена эндотелиальной NO-синтетазы ассоциируется со снижением эндотелийзависимой вазодилатации после манжеточной пробы, что может свидетельствовать о более выраженной эндотелиальной дисфункции у этой категории больных (рис. 2).
/49/49.jpg)
Жесткость сосудистой стенки оценивалась путем измерения скорости распространения пульсовой волны на участках сонной — бедренной и сонной — лучевой артерий неинвазивным методом (аппарат Complior®). СРПВ на участке сонной — лучевой артерий в большей степени отражает изменения жесткости в артериях мышечного типа, а на участке сонной — бедренной артерий — в артериях эластического типа. Обследовано 73 больных с ОКС. По основным клинико-анамнестическим характеристикам, а также частоте распределения различных вариантов генотипа промотора гена eNOS данная выборка больных соответствовала общей совокупности обследованных пациентов. Распределение –786ТТ, –786ТС и –786СС генотипов соответствовало 41,1; 46,6 и 12,3 %.
/49/49_2.jpg)
Пациенты с генотипом СС промотора гена eNOS характеризовались достоверно более высокой СРПВ на участке как сонной — лучевой, так и сонной — бедренной артерий по сравнению с таковой у больных с генотипом ТТ. СРПВ у больных с нормальным (–786ТТ) генотипом и гетерозигот (–786ТС) по данному генотипу не отличались, однако суммарно носители измененной С-аллели (–786ТС и –786СС) характеризовались более высокой СРПВ на участке сонной — лучевой артерий (табл. 4).
Оксид азота (NO) не только активно участвует в регуляции тонуса сосудистой стенки, ее проницаемости для липидов и клеток крови, интенсивности клеточной пролиферации, но и контролирует активность внутрисосудистого тромбообразования и фибринолиза [4, 17, 32]. Нами проведена оценка влияния полиморфизма промотора гена eNOS на эффективность тромболитической терапии у больных с острым ИМ.
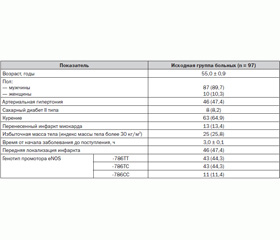
Отобрано 97 пациентов с ОКС со стойкой элевацией сегмента ST на ЭКГ, которым проводилась системная фибринолитическая терапия путем внутривенной инфузии стрептокиназы в дозе 1,5 млн МЕ в течение 30–45 минут. Среднее время от начала болевого синдрома до госпитализации в стационар составило 3,0 ± 0,1 часа, а время до начала проведения тромболитической терапии — 3,5 ± 0,1 часа. В данной выборке больных частота регистрации различных вариантов генотипа промотора гена eNOS соответствовала таковой в общей группе больных ОКС с элевацией сегмента ST (–786ТТ/–786ТС/–786СС как 44,3/44,3/11,4 %).
Исходная характеристика обследованных больных представлена в табл. 5.
/50/50.jpg)
Больные с различным генотипом промотора гена eNOS не отличались по основным клинико-анамнестическим характеристикам, однако в группе пациентов с –786СС генотипом достоверно чаще выявлялись женщины и больные с избыточной массой тела (табл. 5).
При анализе динамики вымывания МВ-фракции КФК отмечено, что больные с –786ТТ генотипом характеризовались достоверно более высоким пиковым уровнем данного фермента, чем у пациентов с полиморфизмом гена eNOS. У этой категории больных были отмечены также более быстрое достижение пиковых значений и более ранняя нормализация уровня МВ-КФК (табл. 6). Быстрое вымывание МВ-фракции КФК указывает на наличие восстановленного коронарного кровотока и отсутствие дальнейшего повреждения жизнеспособных кардиомиоцитов, а также свидетельствует о стабильности зоны некроза за наблюдаемый период. По данным ряда авторов, продолжительная гиперферментемия является маркером увеличения зоны некроза или возникновения дополнительных очагов поражения в сердечной мышце [1]. В первые 12 часов от начала болевого синдрома пик МВ-КФК был достигнут у 30 (69,8 %) больных с –786ТТ генотипом, 18 (41,9 %) больных с –786ТС генотипом и у 4 (36,4 %) больных с –786СС генотипом. Полученные данные свидетельствуют, что открытие ИОКА достоверно более часто регистрировалось у больных с –786ТТ генотипом, чем у больных с полиморфизмом промотора гена eNOS. Данные результаты подтверждались также другими неинвазивными маркерами открытия ИОКА (наличие реперфузионных аритмий, динамика сегмента ST на электрокардиограмме, купирование болевого синдрома). При этом больные выделенных групп не отличались между собой по частоте использования основных групп антикоагулянтных, антитромбоцитарных и антиишемических препаратов как при проведении тромболитической терапии, так и в течение всего госпитального периода заболевания. Таким образом, в нашем исследовании –786ТТ генотип промотора гена eNOS ассоциировался с более частым открытием ИОКА при проведении тромболитической терапии у больных с ОКС со стойкой элевацией сегмента ST на электрокардиограмме, чем у больных с полиморфизмом гена eNOS.
Проведена оценка течения госпитального периода заболевания у больных с ОКС в зависимости от генотипа промотора гена eNOS. Отмечена тенденция к более частому развитию желудочковых нарушений ритма у больных с –786ТТ генотипом. Подобное увеличение регистрации желудочковой экстрасистолии и идиовентрикулярного ритма связано с большей эффективностью тромболитической терапии у этой категории больных и является следствием реперфузионного повреждения миокарда. Однако по частоте развития жизнеопасных аритмий (желудочковая тахикардия и фибрилляция желудочков), а также блокад сердца достоверных отличий между группами получено не было. Вероятность развития других осложнений в течение первых суток заболевания у больных выделенных групп также достоверно не отличалась (табл. 7).
/51/51.jpg)
Частота внутригоспитальных осложений (после 1-х суток) была выше у больных с полиморфизмом гена eNOS. В этой группе (носители измененной аллели С — суммарно –786ТС и –786СС генотипы) достоверно чаще регистрировалась желудочковая экстрасистолическая аритмия на 10-е сутки и была отмечена тенденция к более частой регистрации ОЛЖН (Killip II–III) на 5-е сутки от развития ИМ. Не было выявлено статистически достоверных различий между группами по частоте развития смерти, нефатального инфаркта миокарда и острого нарушения мозгового кровообращения (ОНМК). Однако у больных с наличием генетического полиморфизма более часто отмечалось развитие комбинированной конечной точки «смерть/нефатальный ИМ/ОНМК», а также ранней постинфарктной стенокардии, что имело статистическую силу в виде тенденции.
Таким образом, больные с ОКС со стойкой элевацией сегмента ST и –786ТТ генотипом промотора гена eNOS, которым проводилась терапия стрептокиназой с целью реперфузии, характеризуются более благоприятным течением госпитального периода заболевания, что может быть связано как с большей эффективностью фибринолитической терапии, так и с другими NO-зависимыми механизмами.
Обсуждение
В эксперименте было показано, что наличие аллели С в положении 786 промотора гена eNOS приводит к снижению его активности на 52 ± 11 %, что было впоследствии подтверждено в исследовании in vivo [13]. Формирующийся в результате уменьшения экспрессии гена недостаток eNOS является причиной уменьшения синтеза и высвобождения оксида азота и дисфункции эндотелия [36, 39, 40, 42, 44–46]. В нашем исследовании обнаружено, что полиморфизм гена эндотелиальной NO-синтетазы ассоциируется со снижением эндотелийзависимой вазодилатации после манжеточной пробы, что подтверждает наличие эндотелиальной дисфункции у этой категории больных. Нами также впервые продемонстрирована связь между полиморфизмом гена eNOS и жесткостью артерий эластического и мышечного типа. Этот показатель в настоящее время используется как интегральный маркер состояния сосудистой стенки, поэтому его повышение у больных с полиморфизмом NO свидетельствует о более выраженных и длительных структурных и функциональных изменениях сосудистой системы у данной категории больных. Эти изменения в клинике могут реализоваться увеличением тонуса венечных артерий, повышенной склонностью к коронароспазму и извращенной реакции венечных артерий на введение ацетилхолина, повышенным риском рестенозов после стентирования венечных артерий, что было описано в литературе [15, 26, 33, 35, 36, 47, 48]. В дополнение к своей общеизвестной роли в регуляции сосудистого тонуса оксид азота также предупреждает агрегацию тромбоцитов, повышает высвобождение тканевого активатора плазминогена, является модулятором активности симпатического и парасимпатического звеньев вегетативной нервной системы [3, 4, 16, 36, 39, 44]. Приведенные данные литературы и результаты нашего исследования свидетельствуют о более выраженной сосудистой и автономной дисфункции у больных с полиморфизмом гена eNOS и таким образом определяют категорию больных с высоким риском развития осложнений.
Нами впервые (по данным доступной литературы) выполнена фармакогенетическая оценка эффективности проведения тромболитической терапии в зависимости от генотипа промотора гена eNOS. У больных с ОКС со стойкой элевацией сегмента ST на электрокардиограмме показано, что –786ТТ генотип промотора гена eNOS ассоциируется с более частым открытием ИОКА при проведении тромболитической терапии. Объяснением данного факта может служить связь между функцией эндотелия и высвобождением тканевого активатора плазминогена. Было показано, что внутриартериальная инфузия ацетилхолина (агонист эндотелия) вызывает быстрое высвобождение тканевого активатора плазминогена, а блокада эндотелиальной NО-синтетазы (NG-монометил — L-аргинин) приводит к значительному снижению его уровня. NO также является активным регулятором процесса тромбообразования: при агрегации тромбоцитов включается NO-зависимый механизм отрицательной обратной связи, ограничивающий этот процесс. Первое звено данного механизма состоит в высвобождении NO, ингибирующего агрегацию и адгезию, из самих агрегирующих тромбоцитов. Второе звено представляет собой взаимодействие между тромбоцитами и эндотелиальными клетками. Агрегирующие тромбоциты высвобождают АДФ, который активирует эндотелиальную NO-синтазу (eNOS). Образующийся эндотелиальный NO предупреждает агрегацию тромбоцитов и противодействует вазоконстрикторному эффекту тромбоксана А2 и серотонина, которые продуцируются тромбоцитами. В условиях дефицита эндотелиального NO этот защитный механизм не работает, и, соответственно, создаются условия, способствующие вазоконстрикции, тромбозам и ишемии. Считается также, что дисфункция эндотелия приводит к снижению уровня тромбомодулина — белка, который блокирует способность тромбина расщеплять фибриноген и активирует протеин С [16]. Описанные выше патогенетические механизмы, по всей вероятности, явились причиной более благоприятного течения острого периода ИМ у больных с –786ТТ генотипом промотора гена eNOS.
Выводы
1. Встречаемость генотипа –786СС промотора гена эндотелиальной NO-синтетазы в украинской популяции достоверно выше, чем в японской, и меньше, чем у западноевропейцев, белых северо–американцев и австралийцев.
2. Генотип –786СС промотора примерно в 3,5 раза чаще встречается у больных с острым коронарным синдромом, чем у здоровых лиц. Полученные данные указывают на патогенетическое значение данного полиморфизма в развитии острых форм ишемической болезни сердца, особенно у мужчин с преждевременным развитием атеросклероза (до 55 лет).
3. Полиморфизм гена эндотелиальной NO-синтетазы ассоциируется со снижением эндотелийзависимой вазодилатации и повышенной жесткостью артерий мышечного и эластического типа у больных с ОКС, что может указывать на возможные механизмы реализации измененного генотипа.
4. У больных с ОКС со стойкой элевацией сегмента ST на электрокардиограмме –786ТТ генотип промотора гена eNOS ассоциируется с более частым открытием инфарктобусловливающей коронарной артерии при проведении тромболитической терапии стрептокиназой, а также более благоприятным течением госпитального периода заболевания.
1. Гватуа Н.А., Солоненко И.Н., Шкляр Л.В. Особенности формирования зоны некроза при крупноочаговом и мелкоочаговом инфаркте миокарда // Кардиология. — 1988. — № 11. — С. 112–114.
2. Досенко В.Є., Загорій В.Ю., Мойбенко О.О., Пархоменко О.М. Патофізіологічні аспекти генетичного поліморфізму ендотеліальної NO–синтази // Фізіол. журн. — 2002. — Т. 48, № 6. — С. 86–102.
3. Мойбенко О.О., Павлюченко В.Б., Даценко В.В. та ін. Дослідження ролі ендотелійзалежних факторів у реалізації кардіогенних рефлексів за нормальних та патологічних умов // Фізіол. журн. — 2000. — Т. 46, № 2. — С. 19–32.
4. Мoйбенко О.О., Сагач В.Ф., Шаповал Л.М. та ін. Роль ендотелію та біологічно-активних речовин ендотеліального походження в регуляції кровообігу та діяльності серця // Фізіол. журн. — 1997. — Т. 43, № 1–2. — С. 3–18.
5. Alvarez R., Gonzalez P., Batalla A. et al. Association between the NOS3 (–786 T/C) and the ACE (I/D) DNA genotypes and early coronary artery disease // Nitric. Oxide. — 2001. — Vol. 5, № 4. — P. 343–348.
6. Asmar R., Topouchian J., Pannier B. et al. Pulse wave velocity as endpoint in large-scale intervention trial (The Complior Study) // J. Hypertens. — 2001. — 19. — 813–18.
7. Bertrand M.E., Simoons M.L., Fox K.A.A. et al. Management of acute coronary syndromes in patients presenting without persistent ST-segment elevation. The Task Force on the Management of Acute Coronary Syndromes of the European Society of Cardiology // Eur. Heart J. — 2002. — Vol. 23. — P. 1809–1840.
8. Braunwald E., Antman E.M., Brooks N.H. et al. ACC/AHA guidelines for the management of patients with unstable angina and non ST-elevation myocardial infarction: Executive summary and Recommendations. A report of the American College of Cardiology/ American Heart Association task force on practice guidelines (Committee on management of patients with unstable angina) // Circulation. — 2000. — Vol. 102. — P. 1193–1209.
9. Bredt D.S., Hwang P.M., Glatt C.E., Lowenstein C. et al. Cloned and expressed nitric oxide synthase structurally resembles cytochrome P-450 reductase // Nature. — 1991. — Vol. 351. — P. 714–718.
10. Casas J.P., Bautista L.E., Humphries S.E., Hingorani A.D. Endothelial nitric oxide synthase genotype and ischemic disease. Meta-analysis of 26 studies involving 23028 subjects // Circulation. — 2004. — Vol. 109. — P. 1359–1365.
11. Colombo M.G., Paradossi U., Andreassi M.G. et al. Endothelial nitric oxide synthase gene polymorphisms and risk of coronary artery disease // Clin. Chem. — 2003. — Vol. 49. — P. 389–395.
12. Corretti M, Anderson T., Benjamin E. et al. Guidelines for the Ultrasound Assessment of Endothelial-Dependent Flow-Mediated Vasodilatation of the Brahial Artery // J. Am. Coll. Cardiol. — 2002. — Vol. 39. — P. 257–265.
13. Doshi A., Lesinski A., Binkley P. A Promoter Polymorphism of the Endothelial Nitric Oxide Synthase Gene Reduces Endothelial Nitric Oxide Synthase Expression in Patients With Heart Failure // Circulation. — 2006. — Vol. 114. — P. 802.
14. Ghilardi G., Biondi M.L., DeMonti M. et al. Independent risk factor for moderate to severe internal carotid artery stenosis: T786C mutation of endothelial nitric oxide synthase gene // Clin. Chem. — 2002. — Vol. 48, № 7. — P. 989–993.
15. Gomma A.H., Elrayess M.A., Knight C.J. et al. The endothelial nitric oxide synthase (Glu298Asp and –786T > C) gene polymorphisms are associated with coronary in-stent restenosis // Eur. Heart J. — 2002. — Vol. 23. — P. 1955–1962.
16. Gregory Y.H. Effects of hormone-replacement therapy on hemostatic factors, lipid factors and endothelial functions in women undergoing surgical menopause: implications for prevention for atherosclerosis // Am. Heart J. —1997. — Vol. 134(4). — P. 164–771.
17. Harrison D.G. Cellular and molecular mechanisms of endothelial cell dysfunction // J. Clin. Invest. — 1997. — Vol. 19. — P. 23–27.
18. Hingorani A.D. Polymorphisms in endothelial nitric oxide synthase and atherogenesis: John French Lecture 2000 // Atherosclerosis. — 2001. — Vol. 154. — P. 521–527.
19. Hingorani A.D., Jia H., Stevens P.A. et al. A common variant in exon 7 of the endothelial constitutive nitric oxide synthase gene // Clin. Sci. — 1995. — Vol. 88. — P. 21.
20. Hohnloser S.H., Label M., Kasper W. et al. Assessment of coronary artery patency after thrombolytic therapy: accurte prediction utilizing the combined analysis of three noninvasive markers // J. Am. Coll. Cardiol. — 1991. — Vol. 18, № 1. — P. 44–49.
21. Hyndman M.E., Parsons H.G., Verma S. et al. The T–786®C mutation in endothelial nitric oxide synthase is associated with hypertension // Hypertension. — 2002. — Vol. 39. — P. 919–925.
22. Iwai N., Katsuya T., Ishikawa K. et al. Human prostacyclin synthase gene and hypertension: the Suita Study // Circulation. — 1999. — Vol. 100. — P. 2231–2236.
23. Janssens S.P., Shimouchi A., Quetermous T. et al. Cloning and expression of cDNA encoding human endothelium-derived relaxing factor/nitric oxide synthase // J. Biol. Chem. — 1992. — Vol. 267. — P. 14519–14522.
24. Jeerooburkhan N., Jones L.C., Bujac S. et al. Genetic and environmental determinants of plasma nitrogen oxides and risk of ischemic heart disease // Hypertension. — 2001. — Vol. 38. — P. 1054–1061.
25. Koenig W. Atherosclerosis involves more than just lipids: focus on inflammation // Eur. Heart J. — 1999. — Vol. 1 (suppl. T). — P. 19–26.
26. Kugiyama K., Yasue H., Ohgushi M. et al. Deficiency in nitric oxide bioactivity in epicardial coronary arteries of cigarette smokers // J. Amer. Coll. Cardiology. — 1996. — Vol. 28. — P. 1161–1167.
27. Lamas S., Marsden P.A., Li G.K. et al. Endothelial nitric oxide synthase: molecular cloning and characterization of a distinct constitutive enzyme isoform // Proc. Natl. Acad. Sci. USA. — 1992. — Vol. 89. — P. 6348–6352.
28. Lowenstein C.J., Glatt C.E., Bredt D.S., Snyder S.H. Cloned and expressed macrophage nitric oxide synthase contasts with the brain enzyme // Proc. Natl. Acad. Sci. USA. — 1992. — Vol. 89. — P. 6711–6715.
29. Luscher T.F., Tschudi M.R., Wenzel R.R., Noll G. Endotheliale dysfunktion und stickst off monoxid (NO; Nitric Oxide) // Internist. — 1997. — Vol. 38. — P. 411–419.
30. Marsden P.A., Heng H.H., Scherer S.W. et al. Structure and chromosomal localization of the human constitutive endothelial nitric oxide synthase gene // J. Biol. Chem. — 1993. — Vol. 268. — P. 17478–17488.
31. Marsden P.A., Schappert K.T., Chen H.S. et al. Molecular cloning and characterization of human endothelial nitric oxide synthase // FEBS Lett. — 1992. — Vol. 307. — P. 287–293.
32. Moncada S., Palmer R.M.J., Higgs E.A. Nitric oxide: physiology, pathology and pharmacology // Pharmacol. Rev. — 1991. — Vol. 43. — P. 109–142.
33. Motoyama T., Kawano H., Kugiyama K. et al. Endothelium-dependent vasodilation in the brachial artery is impaired in smokers: effect of vitamin C // Amer. J. Physiology. — 1997. — Vol. 273. — P. 1644–1650.
34. Naber C.K., Siffert W., Erbe R., Heusch G. Genetics of human coronary vasomotion // Arch. Mal. Coeur. — 2004. — Vol. 97. — P. 255–260.
35. Naber C.K., Oldenburg O., Frey U. et al. Relevance of the T-786C and Glu298Asp variants in the endothelial nitric oxide synthase gene for cholinergic and adrenergic coronary vasomotor responses in man // Circulation. — 2003. — Vol. 106 (suppl. L). — P. 1042 (Abstract).
36. Nakayama M., Yasue H., Yoshimura M. et al. T-786®C mutation in the 5’-flanking region of the endothelial nitric oxide synthase gene is associated with coronary spasm // Circulation. — 1999. — Vol. 99. — P. 2864–2870.
37. Nakayama M., Yasue H., Yoshimura M. et al. T(–786)®C mutation in the 5’-flanking region of the endothelial nitric oxide synthase gene is associated with myocardial infarction, especially without coronary organic stenosis // Amer. J. Cardiology. — 2000. — Vol. 86, № 6. — P. 628–634.
38. Poirier O., Mao C., Mallet C. et al. Polymorphisms of the endothelial nitric oxide synthase gene — no consistent association with myocardial infarction in the ECTIM study // Eur. J. Clin. Invest. — 1999. — Vol. 29. — P. 284–290.
39. Ross R. Atherosclerosis — an inflammatory disease // New Engl. J. Med. — 1999. — Vol. 340. — P. 115–126.
40. Rossi G.P., Taddei S., Virdis A. et al. The T-786C and Glu298Asp polymorphisms of the endothelial nitric oxide gene affect the forearm blood flow responses of Caucasian hypertensive patients // J. Amer. Coll. Cardiology. — 2003. — Vol. 41. — P. 938–945.
41. Sessa W.C. The nitric oxide synthase family of proteins // J. Vasc. Res. — 1994. — Vol. 31. — P. 131–143.
42. Song J., Yoon Y., Park K.U. et al. Genotype-specific influence on nitric oxide synthase gene expression, protein concentration, and enzyme activity in cultured human endothelial cells // Clin. Chem. — 2003. — Vol. 49, № 6. — P. 847–852.
43. Takagi S., Goto Y., Nonogi H. et al. Genetic Polymorphisms of angiotensin converting enzyme (I/D) and endothelial nitric oxide synthase (T(–788)C) genes in Japanese patients with myocardial infarction // Thromb Haemost. — 2001. — Vol. 86. — P. 1339–1340.
44. Tanus-Santos J.E., Desai M., Deak L.R. et al. Effects of endothelial nitric oxide synthase gene polymorphisms on platelet function, nitric oxide release, and interactions with estradiol // Pharmacogenetics. — 2002. — Vol. 12. — P. 407–413.
45. Tsukada T., Yokoyama K., Arai T. et al. Evidence of association of the ecNOS gene polymorphism with plasma NO metabolite levels in humans // Biochem. Biophys. Res. Commun. — 1998. — Vol. 245. — P. 190–193.
46. Yoon Y., Song J., Hong S.H., Kim J.Q. Plasma nitric oxide concentrations and nitric oxide synthase gene polymorphisms in coronary artery disease // Clin. Chem. — 2000. — Vol. 46, № 10. — P. 1626–1630.
47. Yoshimura M., Nakayama M., Shimasaki Y. et al. A T–786C mutation in the 5’-flanking region of the endothelial nitric oxide synthase gene and coronary arterial vasomotility // Amer. J. Cardiology. — 2000. — Vol. 85. — P. 710–714.
48. Yoshimura M., Yasue H., Nakayama M. et al. Genetic risk factors for coronary artery spasm: significance of endothelial nitric oxide synthase gene T-786C and missense Glu298Asp variants // J. Investig. Med. — 2000. — Vol. 48, № 5. — P. 367–374.
49. Zhang R., Min W., Sessa W.C. Functional analysis of the human endothelial nitric oxide synthase promoter. Sp1 and GATA factors are necessary for basal transcription in endothelial cells // J. Biol. Chem. — 1995. — Vol. 270. — P. 15320–15326.