
Международный эндокринологический журнал 1 (65) 2015
Вернуться к номеру
Комбинированная терапия «метформин + глимепирид» у больных сахарным диабетом 2-го типа (молекулярные механизмы оптимизации реабилитирующего действия)
Авторы: Полторак В.В., Красова Н.С. - ГУ «Институт проблем эндокринной патологии им. В.Я. Данилевского НАМН Украины», г. Харьков; Горшунская М.Ю. - Харьковская медицинская академия последипломного образования
Рубрики: Эндокринология
Разделы: Справочник специалиста
Версия для печати
Обзор посвящен описанию молекулярных механизмов терапевтического действия комбинации пероральных антидиабетических препаратов «метформин + глимепирид», направленного на коррекцию обоих дефектов, которые определяют развитие метаболических нарушений при сахарном диабете 2-го типа, а именно относительного дефицита инсулина и инсулинорезистентности. Сочетание метформина с глимепиридом оптимально в плане терапевтического воздействия на обе молекулы, недостаточная активность которых определяет метаболическое состояние инсулинорезистентности, соответственно аденозинмонофосфатактивируемую протеинкиназу и пролифератором пероксисом активированный рецептор γ. Кроме того, метформину присуща уникальная способность устранять феномен «гипергликемической памяти» за счет повышения экспрессии и активности сиртуина-1. С другой стороны, препарат сульфонилмочевины третьей генерации глимепирид обеспечивает «щадящую» стимуляцию β-клеток, что сохраняет физиологическое соотношение проинсулин/инсулин и замедляет развитие феномена десенситизации β-клеток к физиологическим и фармакологическим стимулам. Глимепирид сохраняет защитный феномен ишемического прекондиционирования и расширяет спектр терапевтического влияния на патофизиологические составляющие инсулинорезистентности за счет инсулинмиметического и инсулинсенситайзерного эффекта, а также потенцирует благоприятные эффекты метформина (в частности, через адипонектин). С этим связано достижение оптимизированной терапевтической эффективности фармкомбинации при снижении побочного действия (уменьшение дозы глимепирида, метформина).
Огляд присвячено опису молекулярних механізмів терапевтичної дії комбінації пероральних антидіабетичних препаратів «метформін + глімепірид», спрямованої на корекцію обох дефектiв, які визначають розвиток метаболічних порушень за цукрового діабету 2-го типу, а саме відносного дефіциту інсуліну та інсулінорезистентності. Поєднання метформіну з глімепіридом оптимально в плані терапевтичного впливу на обидві молекули, недостатня активність яких визначає метаболічний стан інсулінорезистентності, відповідно аденозинмонофосфатактивовану протеїнкіназу та проліфератором пероксисом активований рецептор γ. Крім того, метформіну притаманна унікальна здатність усувати феномен «гіперглікемічної пам’яті» за рахунок підвищення експресії та активності сиртуїну-1. З іншого боку, препарат сульфонілсечовини третьої генерації глімепірид забезпечує «щадну» стимуляцію β-клітин, що зберігає фізіологічне співвідношення проінсулін/інсулін і уповільнює розвиток феномена десенситизації β-клітин до фізіологічних і фармакологічних стимулiв. Глімепірид зберігає захисний феномен ішемічного прекондиціювання і розширює спектр терапевтичного впливу на патофізіологічні складові інсулінорезистентності за рахунок інсулінміметичного та інсулінсенситайзерного ефекту, а також потенціює сприятливі ефекти метформіну (зокрема, через адипонектин). З цим пов’язано досягнення оптимізованої терапевтичної ефективності фармкомбінації при зниженні побічної дії (зменшення дози глімепіриду, метформіну).
This review describes the molecular mechanisms of therapeutic action of the combination of oral antidiabetic drugs «metformin + glimepiride», aimed at the correction of both defects, which determine the development of metabolic abnormalities in type 2 diabetes mellitus, namely, relative insulin deficiency and insulin resistance. The combination of metformin with glimepiride is optimal in terms of therapeutic effects on both molecules, which metabolic hypoactivity determines the metabolic state of insulin resistance, respectively, adenosine monophosphate-activated protein kinase and peroxisome proliferator-activated receptor γ. In addition, metformin has unique ability to eliminate the phenomenon of «hyperglycemic memory» by increasing the expression and activity of sirtuin-1. On the other hand, the third generation sulfonylurea glimepiride provides a «gentle» stimulation of the β-cells, which maintains the physiological ratio proinsulin/insulin and slows down the development of the phenomenon of β-cell desensitization to physiological and pharmacological stimuli. Glimepiride retains protective phenomenon of ischemic preconditioning and extends the range of therapeutic effect on the pathophysiological components of insulin resistance due to insulin-mimetic and insulin-sensitizing effect, as well as potentiates the beneficial effects of metformin (particularly, via adiponectin). Above mentioned determinated the achievement of optimized therapeutic efficacy of pharmaceutical combination against the background of decreased side effects (reduced dose of glimepiride, metformin).
сахарный диабет 2-го типа, метформин, глимепирид.
цукровий діабет 2-го типу, метформін, глімепірид.
type 2 diabetes mellitus, metformin, glimepiride.
Статья опубликована на с. 87-97
Сахарный диабет (СД) представляет собой существенную проблему мирового здравоохранения в связи с его высокой глобальной распространенностью, неуклонным ростом и ассоциированными сосудистыми осложнениями. В настоящее время свыше 350 млн человек имеют диагноз СД 2–го типа, и, по прогнозам Международной диабетической федерации, количество больных возрастет до 600 млн к 2035 году. Кроме того, по опубликованным данным, диабет и связанные с ним коморбидности явились причиной 5,1 млн смертей в 2013 году [34, 35].
Патофизиология развития СД 2–го типа характеризуется инсулинорезистентностью и прогрессирующим нарушением секреции инсулина вследствие дисфункции панкреатических b–клеток.
Молекулярные причины инсулинорезистентности, а именно нарушенный инсулиновый сигналинг через фосфатидил–инозитол–3–киназу (метаболический путь) с сохраненной передачей сигнала через митогенактивированные протеинкиназы (митотический путь), являются основой для того, что инсулинорезистентность проявляется, во–первых, в печени — избыточной продукцией глюкозы в базальном состоянии, несмотря на гиперинсулинемию натощак, и слабым торможением печеночной продукции глюкозы в ответ на инсулин и после еды, а во–вторых, в мышцах — нарушенным транспортом глюкозы, что приводит к постпрандиальной гипергликемии [40]. До тех пор пока панкреатические b–клетки способны поддерживать секрецию инсулина на уровне, достаточном для компенсации инсулинорезистентности, толерантность к глюкозе остается относительно нормальной [13]. С другой стороны, к момeнту манифестации СД 2–го типа потеря b–клеток, по данным Британского проспективного исследования сахарного диабета (UK Prospective Diabetes Study — UKPDS), составляет ≥ 50 %, что обосновывает полезность секретагогов для достижения достаточного гликемического контроля [5, 38].
Прогрессирующее развитие СД 2–го типа приводит к тому, что пациентам, начальная терапия которых включала один пероральный антидиабетический препарат (как правило, метформин или сульфонилмочевину), в конечном счете необходимо последовательное добавление других пероральных препаратов для достижения и поддержания гликемического контроля, поскольку персистентная гипергликемия связана с развитием долгосрочных диабетических осложнений [5, 38], а раннее обеспечение хорошего гликемического контроля препятствует формированию негативной «гипергликемической памяти» и, таким образом, обеспечивает снижение риска развития макроангиопатий [79, 94]. Следует отметить, что в добавление к высокой эффективности комбинированной пероральной терапии по сравнению с монотерапией относительно гликемического контроля у больных СД 2–го типа, снижение доз используемых в комбинации препаратов в сопоставлении с более высокими дозами при монотерапии связано с минимизацией повреждающих побочных эффектов [37, 60].
Комбинированная терапия должна включать препараты с различным механизмом действия, направленным на реабилитацию основных патогенетических дефектов при СД 2–го типа [6, 60]. При этом выбор специфических антидиабетических препаратов определяется их глюкозопонижающей эффективностью, экстрагликемическими эффектами (например, снижение факторов риска кардиоваскулярной болезни, таких как артериальная гипертензия или дислипидемия, благоприятные изменения массы тела, инсулиновой резистентности и инсулинсекреторной активности), которые могут уменьшить долгосрочные осложнения, безопасным профилем, переносимостью, легкостью использования и ценой [57].
Наиболее популярной и наиболее изученной комбинацией, направленной на коррекцию обоих дефектов, определяющих развитие метаболических нарушений при СД 2–го типа, а именно относительного дефицита инсулина и инсулинорезистентности, является комбинация сульфонилмочевины c метформином [72]. У многих больных комбинированная пероральная терапия может быть использована как основной подход на ранних этапах манифестного СД 2–го типа вместе с диетой и физическими нагрузками. Позже, с течением болезни, использование пероральных препаратов может замедлить переход к инсулинотерапии при поддержании гликемического контроля, расширяя таким образом контингент больных, которым показана агрессивная пероральная терапия [60].
Метформин (класс бигуанидов) действует главным образом благодаря уменьшению продукции глюкозы печенью и почками (преимущественночерез глюконеогенез) и повышению чувствительности к инсулину в периферических тканях [37]. При приеме метформина в плацебо–контролируемых исследованиях уровень HbА1с понижался на 1–2 % с низким риском гипогликемий, но несущественным влиянием на постпрандиальную гипергликемию [60]. Также улучшался липидный профиль (снижение уровня триглицеридов и холестерина липопротеинов очень низкой плотности), уменьшалась частота микрососудистых и макрососудистых осложнений диабета [31, 79].
Поскольку прием метформина ассоциирован со снижением/сохранением веса вследствие его анорексигенных эффектов на уровне регуляции экспрессии нейропептида Y в гипоталамусе, то его рассматривают как препарат первой линии для терапии тучных больных СД 2–го типа, по крайней мере при отсутствии противопоказаний к использованию [15, 17, 37, 38, 60].
В результате целого ряда экспериментальных и клинических исследований было обнаружено, что одной из точек реализации терапевтических эффектов метформина является аденозинмонофосфатактивируемая протеинкиназа (АМФПК) — эволюционно консервативный сенсор клеточного энергетического статуса, играющий критическую роль в поддержании системного энергетического баланса, а по последним данным, рассматриваемый в качестве главного метаболического регулятора [12, 41]. Показано, что АМФПК объединяет пищевые и гормональные сигналы в периферических тканях и гипоталамусе. Она опосредует эффекты адипокинов, таких как лептин и адипонектин, в регуляции пищевого поведения, массы тела, а также углеводного и липидного гомеостаза и потребления энергии в целом [12]. Эта серин–треониновая киназа активируется, когда уровень энергии в клетке становится низким (т.е. внутриклеточное соотношение АМФ и аденозинтрифосфата (АТФ) высокое), и, соответственно, ингибируется в условиях избыточного поступления питательных веществ (например, гипергликемия, высокожировая диета, ожирение) [80]. При активации АМФПК передает сигнал на расположенные «ниже» субстраты для восстановления нормальных уровней энергетической обеспеченности клетки, стимулируя те процессы, которые генерирует АТФ (например, окисление жирных кислот), и ингибируя те, которые его используют (например, синтез триглицеридов и белков) (рис. 1).
/89/89.jpg)
В целом активация АМФПК, как физиологическая — в результате голодания или физической активности, так и фармакологическая, улучшает чувствительность к инсулину и глюкозный гомеостаз, существенно снижая липотоксичность. Однако в различных тканях ее эффекты несколько отличаются. Так, в скелетных мышцах активация АМФПК стимулирует захват глюкозы инсулинонезависимым образом, окисление жирных кислот, транслокацию глюкозного транспортера GLUT4 в мембраны клеток и биосинтез митохондрий, ингибируя при этом синтез белка и гликогена [78]. Одновременно с этим в сердечной мышце активация АМФПК стимулирует захват глюкозы, окисление жирных кислот и гликолиз [77]. В печени активация АМФПК стимулирует захват глюкозы, гликолиз и окисление жирных кислот при ингибировании глюконеогенеза, синтеза холестерина, триглицеридов, жирных кислот и белка [74]. В жировой ткани происходит мощная стимуляция окисления жирных кислот и снижение синтеза жирных кислот и липолиза [78]. Кроме того, есть данные, что АМФПК ингибирует секрецию инсулина панкреатическими b–клетками [78].
Фармакокинетические данные свидетельствуют о том, что метформин главным образом активирует АМФПК в печени и, в меньшей степени, в мышцах [28]. Это связано в первую очередь с возможностями достижения локальной эффективной концентрации препарата. Влияние метформина на работу АМФПК других периферических тканей связывают с непрямой активацией в результате упомянутого выше центрального анорексигенного действия этого бигуанида [15].
Необходимо отметить, что адипонектин — гормон и адипокин с доказанным противовоспалительным и инсулинсенситайзерным действием также реализует часть своих благоприятных эффектов, таких как снижение массы тела, улучшение чувствительности к инсулину, повышение окисления жирных кислот и угнетение воспаления, через активацию АМФПК [12, 30].
Еще одной точкой реализации терапевтического влияния метформина, активно исследуемой в настоящее время и тесно связанной (взаимной активацией) с работой АМФПК, является сиртуин–1 — НАД+–зависимая деацетилаза, мишенями которой являются гистоны, транскрипционные факторы, корегуляторы, а также метаболические ферменты, которые адаптируют экспрессию генов и метаболическую активность в ответ на энергетический статус клетки [94]. В настоящий момент выявлено по меньшей мере 34 различных белка–мишени для сиртуина–1, участвующих в таких клеточных процессах, как углеводный/липидный метаболизм, биогенез митохондрий, воспаление, аутофагия, стресс–резистентность, апоптоз, обеспечение циркадных ритмов и «молчание» генов [70].
В последние годы было обнаружено, что даже транзиторная гипергликемия способна вызывать в клетках сосудистого эпителия долговременные активирующие эпигенетические (не связанные с нарушениями в последовательности ДНК) изменения [19]. Данные изменения, относимые к феномену гликемической сосудистой памяти, приводят к повышению синтеза ряда провоспалительных компонентов, таких как хемоаттрактантный белок моноцитов 1 и адгезивные молекулы сосудистых клеток 1, которое сохраняется даже после нормализации гликемии. Было выявлено, что в формировании сосудистой «гипергликемической памяти» ключевую роль играет усиление экспрессии митохондриального адаптора р66Shc, отвечающего за генерирование активных форм кислорода в митохондриях [93, 61], при этом доказано, что именно сиртуин–1 регулирует транскрипцию гена р66Shc, обусловливая необходимость повышения активности этого сиртуина при СД 2–го типа для устранения эффекта гликемической памяти и предупреждения/торможения развития эндотелиальной дисфункции и сосудистых осложнений [42, 62].
Учитывая вышеизложенное, доказанная in vitro и in vivo фармакологическая активация сиртуина–1 (так называемое стирание гликемической памяти) и АМФПК с использованием метформина, в целом имитирующая ограничение калоража, является существенной и необходимой составляющей ранней комплексной терапии больных СД 2–го типа, в том числе с избыточным весом [7, 58].
Вместе с тем в связи с прогрессирующим характером СД 2–го типа (в первую очередь усиление дисфункции b–клеток), а также из–за угнетающего секрецию инсулина влияния активации АМФПК эффективность гликемического контроля с помощью монотерапии метформином со временем закономерно снижается. Tак, в соответствии со ставшими классическими результатами UKPDS после трехлетней монотерапии метформином только у 44 % больных поддерживался целевой уровень HbА1c (меньше 7 %), а спустя 9 лет он был верифицирован лишь у 13 % пациентов [84]. И вполне логичным по мере снижения эффективности метформина является включение в терапевтический арсенал других препаратов с дополнительными механизмами. В целом ряде исследований была показана более высокая эффективность такой комбинированной терапии (сочетание метформина с тиазолидиндионами, акарбозой, сульфонилмочевиной, ситаглиптином) для достижения и поддержания нормального гликемического контроля [23, 37, 77, 85].
С другой стороны, несмотря на то, что инсулинорезистентность является ключевой особенностью СД 2–го типа, сама по себе она не способна приводить к развитию диабета. Длительные клинические исследования показали, что появление выраженной гипергликемии ассоциировано с ухудшением секреторной активности b–клеток [14]. В экспериментах in vitro с перфузируемой поджелудочной железой крыс было обнаружено, что при резком повышении концентрации глюкозы нормальная секреция инсулина имеет двухфазный характер. При этом способность организма формировать первый пик инсулина — так называемая первая (острая) фаза инсулиновой секреции — имеет важное значение для нормальной утилизации глюкозы в постпрандиальный период и для поддержания оптимального глюкозного гомеостаза в целом [14, 40]. Потеря этой способности одновременно с усилением второй (отдаленной) фазы секреции рассматривается как ранний признак дисфункции bклеток или даже как независимый предиктор развития СД 2–го типа и требует специфического терапевтического воздействия [9, 14]. Необходимо отметить, что двухфазная секреция инсулина наблюдается не только в ответ на глюкозу, но и вызывается другими «энергетическими» стимулами. Кроме того, некоторые фармакологические агенты способны активировать первую и/или вторую фазу инсулиновой секреции, однако при этом отмечается зависимость от уровня имеющейся гликемии. Так, показано стимулирующее воздействие большинства производных сульфонилмочевины первой и второй генерации на острую фазу только у нормогликемических лиц [40, 87], тогда как в условиях гипергликемии необходимый эффект наблюдался лишь при использовании глимепирида [44, 45].
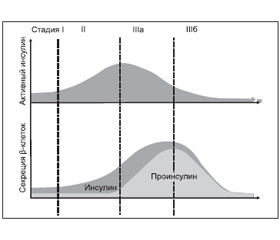
Еще одним показателем дисфункции b–клеток, на котором в настоящее время сфокусировано внимание исследователей, является дефект процессинга («созревания») молекулы проинсулина. Обнаружено, что сниженная секреторная емкость b–клеток приводит к диспропорциональному повышению уровней проинсулина у лиц с нарушенной толерантностью к глюкозе и СД 2–го типа [68]. Была предложена классификация стадий прогрессирования СД 2–го типа, основанная на патофизиологии секреторной активности b–клеток (рис. 2).
/90/90.jpg)
Так, если у инсулинчувствительных пациентов с количественно нормальной секрецией инсулина отсутствует первая фаза инсулинового ответа, то это стадия І (временнóе нарушение) СД. Когда развивается инсулинорезистентность, но b–клетки могут эффективно противодействовать, формируется феномен повышенной секреции активного инсулина (количественное нарушение, стадия ІІ). В дальнейшем клетки могут достигать уровня насыщения процессинговой емкости, и интактный проинсулин секретируется в увеличивающемся количестве (качественное нарушение секреции, стадия ІІІа). В то время как проинсулин является существенной составляющей повышенного кардиоваскулярного риска [39], растущая потребность в активном инсулине может в конечном счете привести к полному истощению секреции b–клеток (стадия ІІІб).
Следует также отметить, что помимо роли проинсулина в качестве информативного биомаркера дисфункции b–клеток в настоящее время верифицирована его важная роль как непрямого предиктора инсулинорезистентности [63] и индивидуального кардиоваскулярного риска [39, 64]. В частности, согласно результатам большого эпидемиологического исследования IRIS–II (4270 человек с СД 2–го типа), увеличение уровней интактного проинсулина было непрямым маркером инсулинорезистентности, а пациенты с повышенными уровнями этого прогормона имели более высокую частоту микро– и макрососудистых осложнений [64]. Результаты 11–летнего Hoorn Study (Нидерланды, 604 участника, в том числе 119 лиц с вновь диагностированным СД 2–го типа) и 27–летнего популяционного наблюдения (Швеция, мужчины первоначально без кардиоваскулярных и онкологических заболеваний, n = 874) показали, что повышенные уровни проинсулина являются фактором риска кардиоваскулярных заболеваний и связанной с ними смертности, не зависящим от других известных факторов кардиоваскулярного риска, в том числе от степени толерантности к глюкозе и инсулинорезистентности [3, 92]. Более того, диспропорциональное увеличение концентрации интактного проинсулина представлялось даже более специфическим маркером инсулинорезистентности и повышенного кардиоваскулярного риска, чем подавление тощаковой адипонектинемии [48]. Атерогенный потенциал проинсулина был подтвержден в двух клинических исследованиях, в которых изучали терапевтический потенциал человеческого проинсулина при СД 2–го типа и обнаружили восьмикратное повышение кардиоваскулярных событий во время лечения человеческим проинсулином по сравнению с человеческим инсулином регуляр [26].
В последние годы поиски патогенетических механизмов запрограммированной гибели панкреатических b–клеток при СД 1–го и 2–го типов привели исследователей к пониманию такого внутриклеточного процесса, как стресс эндоплазматического ретикулума (ЭР–стресс). Эндоплазматический ретикулум — важная клеточная органелла, выполняющая целый ряд функций, включая посттрансляционные модификации и сборку вновь синтезированных секреторных белков, биосинтез липидов и внутриклеточное хранение кальция в депо. Растет количество свидетельств того, что накопление дефектно собранных или медленная деградация мутантных белков являются одними из ведущих причин развития таких патологий, как амилоидоз, фиброз, нейродегенеративные заболевания (прионные болезни, болезнь Альцгеймера и паркинсонизм) и СД 2–го типа [18]. Многие факторы способны нарушать нормальную работу эндоплазматического ретикулума, среди которых при СД 2–го типа выделяют неферментативное гликозилирование белков, снижение образования дисульфидных связей, внезапный выход кальция из депо, нарушение транспорта белков из эндоплазматического ретикулума в аппарат Гольджи, дефекты сборки белков вследствие их избытка и окисления и т.д. Подобная дисфункция вызывает белковую токсичность в эндоплазматическом ретикулуме и при хроническом течении процесса приводит к апоптотической гибели клетки. Поскольку основными функциями панкреатических b–клеток являются синтез и секреция инсулина, эти клетки отличаются чрезвычайно развитым эндоплазматическим ретикулумом и, соответственно, высокой чувствительностью к ЭР–стрессу, весомой составляющей которого является накопление проинсулина [10].
Учитывая вышеуказанное, обоснованное определение уровня интактного проинсулина натощак или отношения проинсулин/инсулин становится распространенным подходом к характеристике состояния b–клеток, связанного с инсулинорезистентностью, и оценке влияния терапевтических вмешательств на инсулинсекретирующие клетки [68].
Показано, что инсулинсенситизирующая терапия (тиоглитазоны, метформин) оказывает благоприятный эффект на функцию b–клеток и процессинг проинсулиновой молекулы [29, 65, 67, 68]. В противоположность инсулинсенсибилизирующей стратегии производные сульфонилмочевины либо не изменяют, либо увеличивают уровни проинсулина плазмы [11, 16, 24, 48, 65–67, 76]. При этом необходимо подчеркнуть обнаруженное нейтральное влияние глимепирида на проинсулинемию и отношение проинсулин/инсулин [65, 69] в противоположность ухудшению (т.е. повышению) этих показателей под воздействием глибенкламида или гликлазида [32]. Имеются, однако, и сообщения об отсутствии влияния на гиперпроинсулинемию и повышенное отношение проинсулин/инсулин у больных СД 2–го типа, получавших глибенкламид [11] или гликлазид [47]. В связи с этим целесообразно еще раз акцентировать внимание на атерогенных эффектах гиперпроинсулинемии и патогенетической обоснованности антидиабетической фармакотерапии, обеспечивающей достижение хорошего метаболического контроля при «щадящей» стимуляции секреции инсулина. «Щадящая» стимуляция b–клеток, обеспечиваемая препаратом сульфонилмочевины третьей генерации глимепиридом, в противоположность персистентной сверхстимуляции препаратами сульфонилмочевины первой и второй генераций, предупреждая/снижая повышенное соотношение проинсулин/инсулин в циркуляции, таким образом уменьшает проатерогенное действие проинсулина. В дополнение меньшее функциональное напряжение b–клеток при терапии глимепиридом (за счет сочетанного инсулинсенситайзерного действия, присущего этому препарату [56, 88]) замедляет/снижает развитие феномена десенситизации инсулиновой секреции, лежащего в основе «вторичной недостаточности производных сульфонилмочевины». Этот феномен определяется как обратимое состояние сниженной секреторной реакции панкреатических b–клеток, вызванное длительной экспозицией последних к множеству стимулов, включая фармакологические средства, инсулинстимулирующeе действие которых связано с деполяризацией и вхождением Са2+ в b–клетку [27, 75]. В частности, показано, что долгосрочная активность b–клеток, индуцируемая толбутамидом, ведет к уменьшению выделяемого инсулина и развитию десенситизации (нечувствительности) инсулиновой секреции, тогда как сигналинговая трансдукция остается без изменения [75]. Вышеизложенное («щадящая» секреция инсулина в отсутствие потенцирующего влияния на качественную дисфункцию b–клеток) обосновывает одно из преимуществ включения глимепирида в комбинированную терапию «метформин + производные сульфонилмочевины» при снижении терапевтической эффективности метформина, связанной со спонтанной эволюцией инсулиновой недостаточности. Кроме того, применение препаратов в комбинации дает дополнительную возможность снизить риск десенситизации и истощения b–клеток благодаря уменьшению эффективной дозы каждого из компо–нентов.
В настоящее время получены веские доказательства того, что глимепирид является оптимальным дериватом сульфонилмочевины для терапии не только нетучных больных СД, у которых нарушена функция панкреатических b–клеток, но и тучных больных диабетом, у которых нарушение чувствительности к инсулину является детерминирующим в патофизиологии процесса [36]. Так, на модели полностью дифференцированных адипоцитов 3Т3–L1 был продемонстрирован плейотропный эффект глимепирида, а именно индукция активности пролифератором пероксисом активируемого рецептора y (РРАRy). Следует сказать, что стимулированная глимепиридом эндогенная транскрипционная активность была отмечена при фармакологически релевантной концентрации глимепирида — 1 мкмоль. В связи с этим интересно подчеркнуть, что минимальная концентрация глибенкламида, стимулирующая активность РРАRy, составила 10 мкмоль, что превышает фармакологические концентрации [36]. По мнению авторов, вышеотмеченнoе может объяснить фармакологические различия между глимепиридом и препаратами сульфонилмочевины второй генерации в физиологических условиях. Исходя из доказанной критической роли РРАRy в адипогенезе и существенного их участия в дифференциации адипоцитов [21], привлекает внимание повышение глимепиридом в 2,4 и 3,7 раза транскрипционных уровней аР2, включенного в дифференциацию адипоцитов 3Т3–L1, соответственно в адипоцитах и фибробластах 3Т3–L1, являющихся, как известно, предшественниками адипоцитов [36]. Эти результаты свидетельствуют о том, что глимепирид способен стимулировать РРАRy–мишеневые гены через повышение активноcти РРАRyin vitro. Очевидно, что это фармакологическое действие глимепирида не зависит от связывания с сульфонилмочевинными рецепторами, поскольку последние не экспрессируются в жировой ткани [1]. В то же время необходимо отметить, что активация РРАRy глимепиридом составляет около 16–25 % от зафиксированной при использовании наиболее известных стимуляторов этой активности — тиазолидиндионов, а именно пиоглитазона [25]. Однако данный факт не следует рассматривать как недостаток, напротив, в настоящее время большинство побочных эффектов, характерных для тиазолидиндионов, связывают с избыточной стимуляцией РРАRy, что приводит к развитию остеопороза (вследствие преимущественного образования адипоцитов вместо остеобластов из общих мезенхимальных прогениторных клеток) и повышению риска инфаркта миокарда (для розиглитазона) и рака мочевого пузыря (для пиоглитазона) [49, 54, 86]. В связи с вышеизложенным привлекают внимание результаты, полученные in vitro на мандибулярных остеобластах крыс, свидетельствующие о позитивном влиянии глимепирида на дифференциацию клеток кости в условиях гипергликемии [51].
Вызывает интерес и способность глимепирида повышать уровень адипонектина у больных СД 2–го типа, также, по всей видимости, благодаря активации РРАRy [36, 46, 83]. Адипонектин является единственным инсулинсенситайзерным гормоном жировой ткани с доказанным клинически превентивным эффектом относительно развития CД 2–го типа и его макрососудистых осложнений [33, 43, 81]. С другой стороны, экспрессия адипонектина регулируется транскрипционной активностью РРАRy, и адипонектин рассматривается как наиболее вероятный ген модуляции чувствительности к инсулину cреди многочисленных мишеневых генов РРАRy [36]. Следует отметить, что другая мишеневая молекула, уменьшающая инсулинорезистентность, а именно упомянутая выше АМФПК, активация которой является ключевой в инсулинсенсибилизирующем действии голодания, бигуанидов и физических упражнений [28], не изменялась в культуре миоцитов даже при высокой концентрации глимепирида (50 мкмоль) [36]. Полученные результаты на молекулярном уровне подтверждают патофизиологическую обоснованность и терапевтическую перспективность пероральной терапии больных СД 2–го типа комбинацией «метформин + глимепирид», обеспечивающей реабилитирующее влияние на обе молекулы, снижение активности которых лежит в основе инсулинорезистентности.
В пользу перспективности применения комбинированной терапии с использованием глимепирида при длительности СД 2–го типа 10 лет и более свидетельствуют недавно опубликованные результаты рандомизированного двойного слепого плацебо–контролируемого перекрестного исследования, проведенного Каролинским институтом (Швеция) [59]. На основе полученных результатов авторы констатируют, что даже при длительном (10–30 лет) течении диабета добавление глимепирида к терапии инсулином и метформином является эффективным в снижении HbA1c (на 0,5 %, что эквивалентно снижению кардиоваскулярного риска на 11,5 %) и дозы экзогенного инсулина (на ≥ 20 %).
Стоит упомянуть, что глимепирид представляет собой антидиабетический сульфонилмочевинный препарат с доказанным отсутствием повреждающего влияния на кардиопротективный феномен ишемического прекондиционирования у больных СД 2–го типа [50]. И хотя до конца не выяснено клиническое значение ишемического прекондиционирования у человека, имеющиеся к настоящему времени доказательства позволяют говорить о полезности этого адаптивного феномена и правомерности его определения как золотого стандарта кардиопротекции [22]. Глимепирид, обладающий уникальным сочетанием инсулинсекретогенных («щадящая» стимуляция) и инсулинсенситайзерных свойств при нейтральном влиянии на вес и низком риске гипогликемий, может быть лучшим выбором у больных СД 2–го типа при наличии кардиоваскулярного риска по сравнению с другими сульфонилмочевинными препаратами в связи с отсутствием повреждающего эффекта в отношении этого защитного феномена [91].
Не исключено, что именно сохранение феномена ишемического прекондиционирования глимепиридом, оставляющим открытыми КАТФ–каналы мембран митохондрий миокарда, которые доминируют в генезе метаболической адаптации к ишемии [82], и объясняет наименьшую смертность при комбинированной терапии во флорентийском исследовании больных СД 2–го типа [52]. У пациентов с СД, недостаточно компенсированным монотерапией метформином, при добавлении глибенкламида, репаглинида или гликлазида годовая смертность была выше, чем у больных, получавших комбинированную терапию «метформин + глимепирид». По результатам недавно опубликованного ретроспективного популяционного исследования, добавление инсулина к потерявшей успешность терапии метформином (n = 2948) приводило к возрастанию риска нефатальных кардиоваскулярных событий и повышению общей смертности по сравнению с группой больных СД 2–го типа с комбинированной терапией «метформин + сульфонилмочевина» (n = 39 990) [73].
Таким образом, для больных СД 2–го типа вследствие спонтанной эволюции заболевания (персистентная инсулинорезистентность в сочетании с неизбежным прогрессированием инсулиновой недостаточности от относительной к абсолютной) со временем характерно снижение эффективности гликемического контроля препаратом первого выбора метформином. Это усиливается также тем, что АМФПК, основной реализатор снижающего инсулинорезистентность действия бигуанидов, тормозит секрецию инсулина. Поэтому нарастающая со временем инсулиновая недостаточность для достижения целевого гликемического контроля требует «щадящей» стимуляции панкреатических b–клеток.
В настоящее время глимепирид представляет собой наиболее оптимальный сульфонилмочевинный препарат для оптимизации комбинированной терапии с метформином, поскольку обеспечивает максимальное соответствие специфических мишеневых эффектов фармкомбинации детерминирующим патогенетическим факторам СД 2–го типа и ассоциированных ангиопатий.
Глимепирид:
— «щадяще» стимулирует секрецию инсулина с воспроизведением ее физиологического биоритма, повышая прежде всего первую фазу секреции гормона;
— обеспечивает снижение атерогенных эффектов (сохранение физиологического соотношения проинсулин/инсулин) и замедляет развитие феномена десенситизации (нечувствительности) b–клеток к физиологическим (гипергликемия, аргинин) и фармакологическим (в первую очередь производные сульфонилмочевины первой и второй генерации) стимулам;
— сохраняет физиологический ответ к физической нагрузке, включая снижение секреции инсулина;
— повышает синтез и секрецию адипонектина — единственного гормона жировой ткани с доказанным антиатерогенным действием;
— обладает прямым (за пределами гликемического контроля) снижающим инсулинорезистентность действием за счет инсулинмиметического и инсулинсенситайзерного эффекта;
— сохраняет защитный феномен ишемического прекондиционирования;
— расширяет спектр реабилитирующего влияния на патофизиологические составляющие инсулинорезистентности, а также потенцирует благоприятные эффекты метформина (в частности, через адипонектин, который является активатором АМФПК — ключевого фермента в реализации снижающего инсулинорезистентность действия метформина). С этим связано достижение оптимизированной терапевтической эффективности фармкомбинации при снижении побочного действия (уменьшение дозы глимепирида, метформина).
Сочетание метформина с глимепиридом оптимально в плане терапевтического воздействия на обе молекулы, недостаточная активность которых определяет метаболическое состояние инсулинорезистентности, соответственно АМФПК и РРАRy.
Терапевтическая оптимальность фармакологической комбинации «метформин + глимепирид» базируется также на уникальной способности метформина устранять феномен «гипергликемической памяти» за счет повышения экспрессии и активности ключевого участника — сиртуина–1, с одной стороны, и на патогенетически обоснованном соответствии реабилитирующих эффектов такого сочетания спонтанной эволюции СД 2–го типа — с другой.
1. Aguilar–Bryan L., Nichols C.G., Wechsler S.W. et al. Cloning of the beta cell high–affinity sulfonylurea receptor: a regulator of insulin secretion // Science. — 1995. — Vol. 268, № 5209. — P. 423–426.
2. Alexanderson E., Alexanderson G., Sierra C. et al. Treatment of endothelial dysfunction with glimepiride + metformin in patients with type 2 diabetes mellitus: evaluation with position emission tomography // Diabetologia. — 2007. — Vol. 50. — P. S483.
3. Alssema М., Dekker J.M., Nijpels G. et al. Proinsulin concentration is an independent predictor of all–cause and cardiovascular mortality. An 11–year follow–up of the Hoorn Study // Diabetes Care. — 2005. — Vol. 28, № 4. — Р. 860–865.
4. American Diabetes Association. Diagnosis and classification of diabetes mellitus // Diabetes Care. — 2012. — Vol. 35, Suppl. 1. — P. S64–S71.
5. American Diabetes Association. Standards of medical care in diabetes — 2013 // Diabetes Care. — 2013. — Vol. 36, Suppl. 1. — P. S11–S66.
6. Basit A., Riaz M., Fawwad A. Glimepiride: evidence–based facts, trends, and observations (GIFTS) // Vasc. Health Risk Manag. — 2012. — Vol. 8. — P. 463–472.
7. Caton P.W., Nayuni N.K., Kieswich J. et al. Metformin suppresses hepatic gluconeogenesis through induction of SIRT1 and GCN5 // J. Endocrinol. — 2010. — Vol. 205. — P. 97–106.
8. Charpentier G., Fleury F., Kabir M. et al. Improved glycaemic control by addition of glimepiride to metformin monotherapy in type 2 diabetic patients // Diabet. Med. — 2001. — Vol. 18. — P. 828–834.
9. Cheng K., Andrikopoulos S., Gunton J.E. First phase insulin secretion and type 2 diabetes // Curr. Mol. Med. — 2013. — Vol. 13. — P. 126–139.
10. Cnop M., Ladriere L., Igoillo–Esteve M. et al. Causes and cures for endoplasmic reticulum stress in lipotoxic β–cell dysfunction // Diabetes Obes. Metab. — 2010. — Vol. 12, Suppl. 2. — P. 76–82.
11. Cooper M.B., Al Majali K., Bailey C.J., Betteridge D.J. Reduced postprandial proinsulinaemia and 32–33 split proinsulinaemia after a mixed meal in type 2 diabetic patients following sensitization to insulin with pioglitazone // Clin. Endocrinol. (Oxf). — 2008. — Vol. 68, № 5. — P. 738–746.
12. Coughlan K.A., Valentine R.J., Ruderman N.B., Saha A.K. AMPK activation: a therapeutic target for type 2 diabetes? // Diabetes Metab. Syndr. Obes. — 2014. — Vol. 7. — P. 241–253.
13. DeFronzo R.A. Pathogenesis of type 2 diabetes mellitus // Med. Clin. North Am. — 2004. — Vol. 88. — P. 787–835.
14. Del Prato S., Tiengo A. The importance of first–phase insulin secretion: implicatopns for the therapy of type 2 diabetes mellitus // Diabetes Metab. Res. Rev. — 2001. — Vol. 17. — P. 164–174.
15. Duan Y., Zhang R., Zhang M. et al. Metformin inhibits food intake and neuropeptide Y gene expression in the hypothalamus // Neural Regen. Res. — 2013. — Vol. 8, № 25. — P. 2379–2388.
16. Dworacka M., Abramczyk M., Winiarska H. et al. Disproportionately elevated proinsulin levels in type 2 diabetic patients treated with sulfonylurea // Int. J. Clin. Pharmacol. Ther. — 2006. — Vol. 44, № 1. — P. 14–21.
17. Eckel R.H., Kahn S.E., Ferrannini E. et al. Obesity and type 2 diabetes: what can be unified and what needs to be individualized? // J. Clin. Endocrinol. Metab. — 2011. — Vol. 96, № 6. — P. 1654–1663.
18. Eizirik D.L., Cardozo A.K., Cnop M. The role for endoplasmic reticulum stress in diabetes mellitus // Endocr. Rev. — 2008. — Vol. 29, № 1. — 42–61.
19. El–Osta A., Brasacchio D., Yao D. et al. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia // J. Exp. Med. — 2008. — Vol. 205. — P. 2409–2417.
20. Eriksson L., Nyström T. Activation of AMP–activated protein kinase by metformin protects human coronary artery endothelial cells against diabetic lipoapoptosis // Cardiovasc. Diabetol. — 2014. — Vol. 13, № 1. — P. 152.
21. Fajas L., Debril M.B., Auwerx J. Peroxisome proliferator–activated receptor–gamma: from adipogenesis to carcinogenesis // J. Mol. Endocrinol. — 2001. — Vol. 27. — P. 1–9.
22. Ferdinandy P., Schulz R., Baxter G.F. Interaction of cardiovascular risk factors with myocardial ischemia/reperfusion injury, preconditioning, and postconditioning // Pharmacol. Rev. — 2007. — Vol. 59. — P. 418–458.
23. Fonseca V., Rosenstock J., Patwardhan R., Salzman A. Effect of metformin and rosiglitazone combination therapy in patients with type 2 diabetes mellitus: a randomized controlled trial // JAMA. — 2000. — Vol. 283, № 13. — P. 1695–1702.
24. Forst T., Wahren J. New tricks by an old dog // Exp. Diabetes Res. — 2008. — Vol. 2008. — P. 384219.
25. Fukuen S., Iwaki M., Yasui A. et al. Sulfonylurea agents exhibit peroxisome proliferator–activated peceptor γ agonistic activity // J. Biol. Chem. — 2005. — Vol. 280, № 25. — P. 23653–23659.
26. Galloway J.A., Hooper S.A., Spradlin C.T. et al. Biosynthetic human proinsulin. Review of chemistry, in vitro and in vivo receptor binding, animal and human pharmacology studies, and clinical trial experience // Diabetes Care. — 1992. — Vol. 15, № 5. — Р. 666–692.
27. Grill V., Bjorklund A. Overstimulation and β–cell function // Diabetes. — 2001. — Vol. 50. — P. S122–S124.
28. Gruzman A., Babai G., Sasson S. Adenosine monophosphate–activated protein kinase (AMPK) as a new target for antidiabetic drugs: a review on metabolic, pharmacological and chemical considerations // Rev. Diabet. Stud. — 2009. — Vol. 6, № 1. — P. 13–36.
29. Hanley A.J., Zinman B., Sheridan P. et al.; Diabetes Reduction Assessment With Ramipril and Rosiglitazone Medication (DREAM) Investigators. Effect of Rosiglitazone and Ramipril on {beta}–cell function in people with impaired glucose tolerance or impaired fasting glucose: the DREAM trial // Diabetes Care. — 2010. — Vol. 33, № 3. — P. 608–613.
30. Hattori Y., Nakano Y., Hattori S. et al. High molecular weight adiponectin activates AMPK and suppresses cytokine–induced NF–κB activation in vascular endothelial cells // FEBS Letters. — 2008. — Vol. 582. — P. 1719–1724.
31. Holman R.R., Paul S.K., Bethel M.A. et al. 10–year follow–up of intensive glucose control in type 2 diabetes // N. Engl. J. Med. — 2008. — Vol. 359. — P. 1577–1589.
32. Inoguchi T., Umeda F., Kakimoto M. et al. Chronic sulfonylurea treatment and hyperglycemia aggravate disproportionately elevated plasma proinsulin levels in patients with type 2 diabetes // Endocr. J. — 2000. — Vol. 47, № 6. — P. 763–770.
33. Inoue T., Kootoka N., Morooka T. et al. High molecular adiponесtin as a predictor of longterm clinical outcome in patients with coronary artery disease // Am. J. Cardiol. — 2007. — Vol. 100. — P. 569–574.
34. International Diabetes Federation. Diabetes Atlas. — 5th ed. — Brussels, Belgium: International Diabetes Federation, 2011.
35. International Diabetes Federation. Diabetes Atlas. — 6th ed. — Brussels, Belgium: International Diabetes Federation, 2013.
36. Inukai K., Watanabe M., Nakashima Y. et al. Glimepiride enhances intrinsic peroxisome proliferator–activated receptor–gamma activity in 3T3–L1 adipocytes // Biochem. Byophys. Res. Commun. — 2005. — Vol. 328. — P. 484–490.
37. Inzucchi S.E. Oral antihyperglycemic therapy for type 2 diabetes: scientific review // JAMA. — 2002. — Vol. 287, № 3. — P. 360–372.
38. Inzucchi S.E., Bergenstal R.M., Buse J.B. et al. Management of hyperglycemia in type 2 diabetes: a patient–centered approach. Position Statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD) // Diabetes Care. — 2012. — Vol. 35. — P. 1–16.
39. Jia E.Z., Yang Z.J., Zhu T.B. et al. Proinsulin is an independent predictor of the angiographical characteristics of coronary atherosclerosis // Cardiology. — 2008. — Vol. 110, № 2. — Р. 106–111.
40. Joslin’s diabetes mellitus / Ed. by C.R. Kahn [et al.]. — 14th ed. — Boston: Lippincott Williams & Wilkins, 2006. — 328 p.
41. Khan B.B., Alquier T., Carling D., Hardie D.G. AMP–activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism // Cell Metab. — 2005. — Vol. 1. — P. 15–25.
42. Kitada M., Koya D. SIRT1 in type 2 diabetes: mechanisms and therapeutic potential // Diabetes Metab. J. — 2013. — Vol. 37. — P. 315–325.
43. Koenig W., Khuseinova N., Baumert J. et al. Serum concentrations of adiponectin and risk of type 2 diabetes mellitus and coronary disease in apparently healthy middle–aged men: results from the 18–year follow up of a large cohort from southern Germany // J. Am. Cardiol. — 2006. — Vol. 48. — P. 1369–1377.
44. Korytkowski M., Thomas A., Reid L. et al. Glimepiride improves both first and second phases of insulin secretion in type 2 diabetes // Diabetes Care. — 2002. — Vol. 25. — P. 1607–1611.
45. Korytkowski M.T. Sulfonylurea treatment of type 2 diabetes mellitus: focus on glimepiride // Pharmacotherapy. — 2004. — Vol. 24. — P. 606–620.
46. Koshiba K., Nomura M., Nakaya Y., Ito S. Efficacy of glimepiride on insulin resistance, adipocytokines, and atherosclerosis // J. Med. Invest. — 2006. — Vol. 53. — P. 87–94.
47. Kubo K. Effect of pioglitazone on blood proinsulin levels in patients with type 2 diabetes mellitus // Endocr. J. — 2002. — Vol. 49, № 3. — P. 323–328.
48. Langenfeld M.R., Forst T., Standl E. et al. IRIS II Study: sensitivity and specificity of intact proinsulin, adiponectin, and the proinsulin/adiponectin ratio as markers for insulin resistance // Diabetes Technol. Ther. — 2004. — Vol. 6, № 6. — P. 836–843.
49. Lee E.J., Marcy T.R. The impact of pioglitazone on bladder cancer and cardiovascular events // Consult. Pharm. — 2014. — Vol. 29, № 8. — P. 555–558.
50. Lee T.M., Chou T.F. Impairment of myocardial protection in type 2 diabetic patients // J. Clin. Endocrinol. Metab. — 2003. — Vol. 88, № 2. — P. 531–537.
51. Ma P., Xiong W., Liu H. et al. Extrapancreatic roles of glimepiride on osteoblasts from rat mandibular bone in vitro: regulation of cytodifferentiation through PI3–kinases/Akt signalling pathway // Arch. Oral Biol. — 2011. — Vol. 56, № 4. — P. 307–316.
52. Mannucci E., Monami M., Masotti G. et al. All–cause mortality in diabetic patients treated with combination of sulfonylureas and biguanides // Diabetes Metab. Res. Rev. — 2004. — Vol. 20. — P. 44–47.
53. Marquezine G.F., Wajchenberg B.L. Molecular Activity of Insulin and Selective Insulin Resistance // Endocrinologist. — 2007. — Vol. 17. — P. 351–356.
54. Mazziotti G., Canalis E., Giustina A. Drug–induced osteoporosis: mechanisms and clinical implications // Am. J. Med. — 2010. — Vol. 23, № 10. — P. 877–884.
55. Mocanu M.M., Мaddock H.L., Baxter G.F. et al. Glimepiride, a novel sulfonylurea, does not abolish myocardial protection afforded by either ischemic preconditioning or diazoxide // Circulation. — 2001. — Vol. 103, № 25. — P. 3111–3116.
56. Mori R.C., Hirabara S.M., Hirata A.E. et al. Glimepiride as insulin sensitizer: increased liver and muscle responses to insulin // Diabetes Obes. Metab. — 2008. — Vol. 10, № 7. — P. 596–600.
57. Nathan D.M., Buse J.B., Davidson M.B. et al. Medical management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy: a consensus statement of the American Diabetes Association and the European Association for the Study of Diabetes // Diabetes Care. — 2009. — Vol. 32. — P. 193–203.
58. Nelson L.E., Valentine R.J., Cacicedo J.M. et al. A novel inverse relationship between metformin–triggered AMPK–SIRT1 signaling and p53 protein abundance in high glucose–exposed HepG2 cells // Am. J. Physiol. Cell Physiol. — 2012. — Vol. 303. — P. C4–C13.
59. Nyback–Nakell A., Adamson U., Lins P.E., Landtedt–Hallin L. Adding glimepiride to insulin + metformin in type 2 diabetes of more than 10 years’ duration — a randomised, double–blind, placebo–controlled, cross–over study // Diabetes Res. Clin. Pract. — 2014. — Vol. 103. — P. 286–291.
60. O’Keefe J.H., Abuannadi M., Lavie C.J., Bell D.S.H. Strategies for optimizing glycemic control and cardiovascular prognosis in patients with type 2 diabetes mellitus // Mayo Clin. Proc. — 2011. — Vol. 86, № 2. — P. 128–138.
61. Paneni F., Mochara P., Akhmedov A. et al. Gene silencing of the mitochondrial adaptor p66(Shc) suppresses vascular hyperglycemic memory in diabetes // Circ. Res. — 2012. — Vol. 111. — P. 278–289.
62. Paneni F., Volpe M., Luscher T.F., Cosentino F. SIRT1, p66(Shc), and Set7/9 in vascular hyperglycemic memory: bringing all the strands together // Diabetes. — 2013. — Vol. 62. — P. 1800–1807.
63. Pfützer A., Kunt T., Hohberg C. et al. Fasting intact proinsulin is a highly specific predictor of insulin resistance in type 2 diabetes // Diabetes Care. — 2004. — Vol. 27, № 3. — P. 682–687.
64. Pfützer A., Kann P.H., Pfützer A.H. et al. Intact and total proinsulin: new aspects for diagnosis and treatment of type 2 diabetes mellitus and insulin resistance // Clin. Lab. — 2004. — Vol. 50, № 9–10. — P. 567–573.
65. Pfützer A., Hohberg C., Lubben G. et al. Pioneer study: PPARgamma activation results in overall improvement of clinical and metabolic markers associated with insulin resistance independent of long–term glucose control // Horm. Metab. Res. — 2005. — Vol. 37, № 8. — P. 510–515.
66. Pfützer A., Lorra B., Abdollahnia M.R. et al. The switch from sulfonylurea to preprandial short–acting insulin analog substitution has an immediate and comprehensive beta–cell protective effect in patients with type 2 diabetes mellitus // Diabetes Technol. Ther. — 2006. — Vol. 8, № 3. — P. 375–384.
67. Pfützer A., Derwahl M., Jacob S. et al. Limitations of the HOMA–B score for assessment of beta–cell functionality in interventional trials–results from the PIOglim study // Diabetes Technol. Ther. — 2010. — Vol. 12, № 8. — P. 599–604.
68. Pfützner A., Forst T. Elevated intact proinsulin levels are indicative of beta–cell dysfunction, insulin resistance, and cardiovascular risk: impact of the antidiabetic agent pioglitazone // J. Diabetes Sci. Technol. — 2011. — Vol. 5, № 3. — P. 784–793.
69. Purnell J.Q., Weyer C. Weight effect of current and experimental drugs for diabetes mellitus: from promotion to alleviation of obesity // Treat. Endocrinol. — 2003. — Vol. 2. — P. 33–47.
70. Purushotham A., Shug T.T., Xu Q. et al. Hepatocyte–specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation // Cell Metab. — 2009. — Vol. 9. — P. 327–338.
71. Rajendran R., Garva R., Kristic–Demonacos M., Demonacos C. SIRTuins: molecular traffic lights in the crossroad of oxidative stress, chromatin remodeling, and transcription // J. Biomed. Biotechnol. — 2011. — Vol. 2011. — P. 1–17.
72. Rodbard H.W., Blonde L., Braithwaite S.S. et al.; AACE Diabetes Mellitus Clinical Practice Guidelines Task Force. American Association of Clinical Endocrinologists medical guidelines for clinical practice for the management of diabetes mellitus // Endocrine Practice. — 2007. — Vol. 13, Suppl. 1. — P. 1–68.
73. Roumie C.L., Greevy R.A., Grijava C.G. et al. Association between intensification of metformin treatment with insulin vs sulfonylureas and cardiovascular events and all–cause mortality among patients with diabetes // JAMA. — 2014. — Vol. 311, № 22. — P. 2288–2296.
74. Ruderman N.B., Carling D., Prentki M., Cacicedo J.M. AMPK, insulin resistance, and the metabolic syndrome // J. Clin. Invest. — 2013. — Vol. 123, № 7. — P. 2764–2772.
75. Rustenbeck I. Desensitization of insulin secretion // Biochem. Pharmacol. — 2002. — Vol. 63. — P. 1921–1935.
76. Smith S.A., Porter L.E., Biswas N., Freed M.I. Rosiglitazone, but not glyburide, reduces circulating proinsulin and the proinsulin: insulin ratio in type 2 diabetes // J. Clin. Endocrinol. Metab. — 2004. — Vol. 89, № 12. — P. 6048–6053.
77. Srivastva R.A., Pinkosky S.L., Filippov S. et al. AMP–activated protein kinase: an emerging drug target to regulate imbalances in lipid and carbohydrate metabolism to treat cardio–metabolic diseases // J. Lipid Res. — 2012. — Vol. 53, № 12. — P. 2490–2514.
78. Steinberg G.R., Kemp B.E. AMPK in health and disease // Physiol. Rev. — 2009. — Vol. 89, № 3. — Р. 1025–1078.
79. Stratton I.M., Adler A.I., Neil H.A. et al., UKPDS Group. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study // BMJ. — 2000. — Vol. 321. — P. 405–412.
80. Sugden M.C., Holness M.J. Metformin, metabolic stress, and mitochondria. Focus on «A novel inverse relationship between metformin–triggered AMPK–SIRT1 signaling and p53 protein abundance in high glucose–exposed HepG2 cells» // Am. J. Physiol. Cell Physiol. — 2012. — Vol. 303. — P. C1–C3.
81. Tabak A.G., Carstensen M., Witte D.R. et al. Adiponectin trajectories before type 2 diabetes diagnosis. Whitehall II study // Diabetes Care. — 2012. — Vol. 35. — P. 2540–2547.
82. Tann M., Miura T., Tsuchida A. et al. Contribution of both sarcolemmal K–ATP and mitochondrial K–ATP channels to infarct size limitation by K–ATP channels openers: differences from preconditioning in the role of sarcolemmal K–ATP channels // Naunyn–Schmiedeberg’s Arch. Pharmacol. — 2001. — Vol. 364. — P. 226–232.
83. Tsunekawa T., Hayashi T., Suzuki Y. et al. Plasma adiponectin plays an important role in improving insulin resistance with glimepiride in elderly type 2 diabetic subjects // Diabetes Care. — 2003. — Vol. 26. — P. 285–289.
84. Turner R.C., Holman R.R. Metformin and risk of cardiovascular disease // Cardiology. — 1999. — Vol. 91, № 3. — P. 203–204.
85. Umpierrez G., Issa M., Vlajnic A. Glimepiride versus pioglitazone combination therapy in subjects with type 2 diabetes inadequately controlled on metformin monotherapy: results of a randomized clinical trial // Curr. Med. Res. Opin. — 2006. — Vol. 22, № 4. — P. 751–759.
86. Van de Vyver M., Andrag E., Cockburn I.L., Fer–ris W.F. Thiazolidinedione–induced lipid droplet formation during osteogenic differentiation // J. Endocrinol. — 2014. — Vol. 223, № 2. — P. 119–132.
87. Wu C.Z., Pei D., Hsieh A.T. et al. Comparison of insulin sensitivity, glucose sensitivity, and first phase insulin secretion in patients treated with repaglinide or gliclazide // Arch. Pharm. Res. — 2010. — Vol. 33. — P. 411–416
88. Xu D.–Y., Zhao S.–P., Huang Q.–X. et al. Effects of Glimepiride on metabolic parameters and cardiovascular risk factors in patients with newly diagnosed type 2 diabetes mellitus // Diabetes Res. Clin. Pract. — 2010. — Vol. 88. — P. 71–75.
89. Yamauchi T., Kamon J., Minokoshi Y. et al. Adiponectin stimulates glucose utilization and fatty–acid oxidation by activating AMP–activated protein kinase // Nat. Med. — 2002. — Vol. 8. — P. 1288–1295.
90. Yunukai K., Watanabe M., Nakashima Y. et al. Glimeperide enhaces intrinsic peroxisome proliferator–activated receptor–gamma activity in 3T3–L1 adipocytes // Biochem. Byophys. Res. Commun. — 2005. — Vol. 328. — P. 484–490.
91. Zeller M., Danchin N., Simon D. et al. Impact of type of preadmission sulfonylureas on mortality and cardiovascular outcomes in diabetic patients with acute myocardial infarction // J. Clin. Endocrinol. Metab. — 2010. — Vol. 95. — P. 4993–5002.
92. Zethelius B., Byberg L., Hales C.N. et al. Proinsulin is an independent predictor of coronary heart disease. Report from a 27–year follow–up study // Circulation. — 2002. — Vol. 105. — P. 2153–2158.
93. Zhou S., Chen H.Z., Wan Y.Z. et al. Repression of P66Shc expression by SIRT1 contributes to the prevention of hyperglycemia–induced endothelial dysfunction // Circ. Res. — 2011. — Vol. 109. — P. 639–648.
94. Полторак В.В., Красова Н.С., Горшунская М.Ю. Сиртуины как перспективная мишень для профилактики и терапии сахарного диабета // Пробл. ендокрин. патології. — 2014. — № 3. — С. 97–104.