
Международный эндокринологический журнал 3 (67) 2015
Вернуться к номеру
Вроджений гіперінсулінізм: можливості сучасної діагностики та лікування
Авторы: Глоба Є.В. — Український науково-практичний центр ендокринної хірургії, трансплантації ендокринних органів і тканин МОЗ України, м. Київ
Рубрики: Эндокринология
Версия для печати
Вроджений гіперінсулінізм (ВГІ) — одна з основних причин розвитку персистуючих гіпоглікемічних станів у дитячому віці. У статті поданий досвід діагностики й лікування пацієнтів із ВГІ. Нами було обстежено 7 дітей з діагнозом ВГІ віком від 1 міс. до 1,5 року. Для постановки діагнозу ВГІ проводилося стандартне клінічне й лабораторне обстеження, а також молекулярно-генетична й інструментальна діагностика (проведення позитронно-емісійної томографії з 18-F-допою). Молекулярно-генетична діагностика підтвердила діагноз ВГІ в 6 дітей. Усі 7 дітей із ВГІ виявилися різною мірою резистентними до консервативної терапії, мали різний ступінь порушення психомоторного розвитку й судоми. З них 6 дітям було проведено оперативне втручання з досягненням нормоглікемії. Усім дітям з гіпоглікеміями й підвищеним чи нормальним рівнем С-пептиду й інсуліну необхідно виконувати генетичну діагностику й позитронно-емісійну томографію з 18-F-допою для уточнення типу ВГІ. Необхідний подальший розвиток генетичної діагностики для пошуку нових генів ВГІ. Оперативне лікування може бути терапією вибору в дітей з ВГІ, особливо при його фокальній формі.
Врожденный гиперинсулинизм (ВГИ) — одна из основных причин развития персистирующих гипогликемических состояний в детском возрасте. В статье представлен опыт диагностики и лечения пациентов с ВГИ. Нами было обследовано 7 детей с диагнозом ВГИ в возрасте от 1 мес. до 1,5 года. Для постановки диагноза ВГИ проводилось стандартное клиническое и лабораторное обследование, а также молекулярно-генетическая и инструментальная диагностика (проведение позитронно-эмиссионной томографии с 18-F-допой). Молекулярно-генетическая диагностика подтвердила диагноз ВГИ у 6 детей. Все 7 детей с ВГИ оказались в разной степени резистентными к консервативной терапии, имели различную степень нарушения психомоторного развития и судороги. Из них шести детям было проведено оперативное вмешательство с достижением нормогликемии. Всем детям с гипогликемиями и повышенным или нормальным уровнем С-пептида и инсулина необходимо проводить генетическую диагностику и позитронно-эмиссионную томографию с 18-F-допой для уточнения типа ВГИ. Необходимо дальнейшее развитие генетической диагностики для поиска новых генов ВГИ. Оперативное лечение может быть терапией выбора у детей с ВГИ, особенно при его фокальной форме.
Congenital hyperinsulinism (CHI) — one of the main causes underlying the development of persistent hypoglycemic conditions in childhood. The article presents the experience of diagnosis and treatment in patients with CHI. We have examined 7 children diagnosed with СHI aged 1 month to 1.5 years. For diagnosing CHI, we have used routine clinical and laboratory investigations, as well as molecular genetic and instrumental diagnostics (18F-DOPA positron emission tomography). Molecular genetic testing confirmed the diagnosis of CHI in 6 children. All 7 children with CHI had various resistance to conservative treatment, had a different degree of psychomotor development and seizures. Of them, 6 children underwent surgery with achievement of normoglycemia. Genetic testing and 18F-DOPA positron emission tomography should be carried out in all children with hypoglycemia and increased or normal levels of C-peptide and insulin to clarify the type of CHI. Further development of genetic diagnostics is necessary to find new CHI genes. Surgical treatment may be a treatment of choice in children with CHI, particularly in its focal form.
вроджений гіперінсулінізм, гіпоглікемія, діти, діагностика й лікування.
врожденный гиперинсулинизм, гипогликемия, дети, диагностика и лечение.
congenital hyperinsulinism, hypoglycemia, children, diagnosis and treatment.
Статья опубликована на с. 166-170
Вступ
Вроджений гіперінсулінізм (ВГІ) — спадкове захворювання, що характеризується неадекватною гіперсекрецією інсуліну бета-клітинами підшлункової залози й призводить до розвитку персистуючих гіпоглікемічних станів.
У літературі описано понад 8 генів, що беруть участь у розвитку ВГІ. Від 40 до 60 % випадків ВГІ пов’язані з дефектами генів KCNJ11 і ABCC8, що кодують білки, які беруть участь у роботі АТФ-залежних калієвих каналів бета-клітин підшлункової залози. Близько 15–20 % випадків пов’язані з активуючими мутаціями в генах GCK і GLUD1, що регулюють внутрішньоклітинний метаболізм глюкози. У літературі також присутні поодинокі описи випадків ВГІ, пов’язаних з дефектами генів HADH, HNF4a, HNF1a, LC16A1, INSR, UCP2 та ін. [1]. Але в 30–40 % усіх випадків ВГІ досі не вдається виявити молекулярно-генетичні дефекти в зазначених генах.
Поширеність ВГІ перебуває в межах від 1 : 30 000 до 1 : 50 000 новонароджених, а в популяціях з високим рівнем близькоспоріднених шлюбів сягає 1 : 2500 новонароджених [2, 3].
ВГІ був вперше описаний як «ідіопатична гіпоглікемія дитячого віку» вченим І. Мак-Куоррі [4] в 1954 р. Надалі ВГІ позначали такими термінами, як, наприклад, «лейцин-чутлива гіпоглікемія», «синдром дисрегуляції бета-клітин», «персистуючі гіперінсулінемічні гіпоглікемії дитячого віку», «нізидіобластоз» та ін. Термін «нізидіобластоз» був введений Г. Лейдло ще в 1938 р. [3].
Нізидіобластоз — це тотальна трансформація протокового епітелію підшлункової залози бета-клітин, що продукують інсулін. На сьогодні доведено, що подібна морфологічна картина в дитячому віці є нормальною й не належить до причин гіперінсулінізму [5].
Основним критерієм діагнозу ВГІ служить визначення рівня інсуліну в плазмі (понад 2,0 мкОд/мл) на момент гіпоглікемії (глюкоза крові < 2,4 ммоль/л у дітей віком понад один рік і < 2,2 ммоль/л — у дітей до року). Також до критеріїв, що підтверджують діагноз ВГІ, відносять: гіпокетотичний характер гіпоглікемій, гіперглікемічну відповідь на введення глюкагону, високий або нормальний рівень С-пептиду на тлі гіпоглікемії, потребу у високих дозах внутрішньовенного введення глюкози (> 8 мг/кг/хв), низькі показники амінокислот (валіну, лейцину) в крові, нормальні показники контрінсулярних гормонів (соматотропного гормону, кортизолу, глюкагону), відсутність ознак об’ємного утворення підшлункової залози (інсуліноми) за даними ультразвукового дослідження (УЗД) і мультиспіральної комп’ютерної томографії [6].
За останні десятиріччя було здійснено прорив в галузі вивчення етіопатогенетичних механізмів ВГІ.
Морфологічно ВГІ поділяють на 3 основні форми: дифузну (за якої уражені всі бета-клітини підшлункової залози), фокальну (якщо вогнище ураження обмежено невеликою ділянкою гіперплазованих клітин, що містять великі ядра) й атипову [7, 8]. Справжньою причиною гіперсекреції інсуліну при ВГІ найчастіше є неадекватна робота АТФ-залежних К-каналів бета-клітин підшлункової залози, що зумовлено молекулярно-генетичними дефектами генів KCNJ11 і ABCC8 [1].
Порушення функції АТФ-залежних К-каналів, а також дефекти регуляції внутрішньоклітинного метаболізму глюкози можуть призводити до розвитку гіперінсулінемічних гіпоглікемічних станів. Найбільш частою причиною ВГІ є інактивуючі мутації генів KCNJ11 і ABCC8 [9–12].
АТФ-залежні калієві канали бета-клітин являють собою октамерні структури, внутрішні відділи яких представлені 4 субодиницями білка Kir6.2, що кодуються геном KCNJ11, а зовнішні — 4 субодиницями білка SUR1, що кодуються геном ABCC8. Дані канали здатні змінювати ступінь поляризації мембрани клітини. Функціональна активність каналів регулюється рівнем внутрішньоклітинних аденінових нуклеотидів. Інактивуючі мутації генів KCNJ11 і ABCC8 викликають закриття даних каналів, що призводить до надлишкового надходження Cа2+ в клітину й гіперсекреції інсуліну [1, 9].
У літературі описані як автосомно-рецесивні, так і автосомно-домінантні мутації зазначених генів. На сьогодні виявлено понад 150 мутацій в гені ABCC8 і 25 мутацій — у гені KCNJ11 [13].
ВГІ, пов’язаний з рецесивними мутаціями в генах KCNJ11 і ABCC8, характеризується тяжким перебігом, раннім дебютом гіпоглікемії і, як правило, не піддається консервативній терапії. Домінантно успадковані форми перебігають м’якше, маніфестують пізніше й у більшості випадків чутливі до терапії діазоксидом [1, 7, 11].
Крім порушень роботи АТФ-залежних калієвих каналів бета-клітин, причинами розвитку ВГІ можуть бути порушення роботи ферментів, що беруть участь у внутрішньоклітинному метаболізмі глюкози. До них відносять глюкокіназу (ген GCK), глутаматдегідрогеназу (кодується геном GLUD1) і 3-гідрокси-ацилКоА-дегідрогеназу (ген HADH).
ВГІ можуть бути як персистуючими (при ВГІ, інсуліномах), так і транзиторними (при синдромі Беквіта — Відемана, Сотоса, діабетичній фетопатії, затяжному неонатальному гіперінсулінізмі). Гіперсекреція інсуліну призводить до утилізації глюкози клітинами інсулінзалежних тканин і в той же час пригнічує продукцію глюкози, вільних жирних кислот і кетонових тіл. Подібний метаболічний ефект інсуліну формує біохімічну основу, що позбавляє пацієнтів з ВГІ як глюкози, так і альтернативних джерел енергії для головного мозку, збільшуючи ризик розвитку неврологічних розладів [14].
Верифікувати фокальні форми ВГІ можна за допомогою молекулярно-генетичної діагностики, оскільки формування фокуса відбувається при успадкуванні батьківської мутації в генах KCNJ11 і ABCC8 і втрати гомозиготності [15]. Візуалізувати фокус можливо за допомогою позитронно-емісійної томографії з 18-флюоро-L-3,4-дигідроксифенілаланіном (ПЕТ з 18-F-допою) [16].
Картина гіпоглікемічного синдрому при ВГІ є вкрай варіативною. Гіпоглікемії можуть мати безсимптомний характер, м’який перебіг, добре піддаватися консервативній терапії. У 40–50 % випадків гіпоглікемічний синдром не вдається лікувати консервативно [17]. Таким пацієнтам необхідне оперативне лікування — субтотальна панкреатектомія, а в разі фокальних форм ВГІ проводиться селективна резекція фокуса, що призводить до повного одужання дитини зі збереженою функціональною активністю підшлункової залози [18].
Матеріали і методи
За останні 2 роки на базі Українського науково-практичного центру ендокринної хірургії, відділу дитячої та підліткової ендокринології нами було обстежено 7 дітей з діагнозом ВГІ віком від одного місяця до 1,5 року. Усім пацієнтам з уперше встановленим діагнозом ВГІ проводили комплекс діагностичних процедур, що включав моніторинг глікемії (портативними глюкометрами); дослідження гормонального профілю з оцінкою рівня інсуліну, С-пептиду, кортизолу, тиреотропного гормону, оцінку рівня амінокислот і ацилкарнітину, лактату й аміаку; розгорнутий біохімічний аналіз крові; УЗД органів черевної порожнини. Вираженість гіпоглікемічного синдрому оцінювали за ступенем потреби в глюкозі (кількість глюкози, що вводиться внутрішньовенно, на 1 кг маси тіла за 1 хв). Діагноз ВГІ встановлювали на підставі підвищення рівня інсуліну плазми й відсутності кетонів у сечі під час гіпоглікемії, а також за відсутності порушень бета-окислення жирних кислот і об’ємних утворень підшлункової залози (за результатами УЗД). Молекулярно-генетичні дослідження генів (KCNJ11 і ABCC8) проводили в молекулярно-генетичній лабораторії (м. Ексетер, Великобританія) стандартними методами. Візуалізацію вогнищ проводили за допомогою ПЕТ з 18-F-допою (Центр гіперінсулінізму, м. Оденсе, Данія).
Результати та їх обговорення
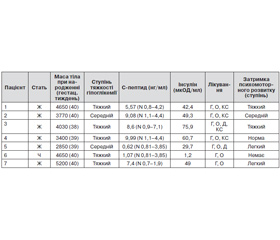
Клінічну характеристику пацієнтів подано в табл. 1. Макросомія спостерігалася в чотирьох пацієнтів (57,1 %), у двох (28,6 %) реєстрували нормальну масу тіла, в однієї дитини (14,3 %) — низьку масу тіла при народженні (відповідно до гестаційного віку). Усі діти мали гіпоглікемії середнього або тяжкого ступеня, у 5 з них був підвищений рівень С-пептиду та інсуліну натще (71,4 %). У двох пацієнтів рівні С-пептиду та інсуліну натще й під час гіпоглікемії були в межах референтних показників.
/168/168.jpg)
Усім дітям лікування розпочинали із введення глюкози внутрішньовенно із швидкістю понад 8 мг/кг/хв, а також у деяких випадках вводився глюкагон внутрішньом’язово (0,5–1,0 мг). Оскільки за таких умов стійкої нормоглікемії досягнуто не було, у чотирьох пацієнтів було розпочато лікування із введенням кортикостероїдів внутрішньовенно (у деяких випадках — до 20 мг/кг маси тіла), потім лікування було змінено на ін’єкції октреотиду (в дозі від 5 до 20 мкг/кг/добу). У двох дітей з дифузною формою досягти нормоглікемії практично не вдавалося, тому дитині з компаундною мутацією p.Q444H/p.Q923X проводили комбіноване лікування за допомогою трьох препаратів (гідрокортизон, октреотид, діазоксид), а дитині без генетично підтвердженого діагнозу ВГІ — комбіноване лікування двома препаратами (октреотид, діазоксид). Усі діти були резистентні до медикаментозної терапії, що проявлялося практично щоденними гіпоглікеміями (в деяких випадках — часті гіпоглікемічні коми з розвитком судом і необхідністю госпіталізації до відділення інтенсивної терапії), а також затримкою психомоторного розвитку різного ступеня тяжкості.
Після встановлення діагнозу всім дітям проводили молекулярно-генетичне обстеження в молекулярно-генетичній лабораторії (м. Ексетер, Великобританія). У шести дітей (85,7 %) було підтверджено генетичну природу ВГІ.
За характером мутації в чотирьох пацієнтів було запідозрено фокальну форму ВГІ (оскільки вони мали мутацію, успадковану від батька). В одного пацієнта з компаундною мутацією (ABCC8, p.Q444H/p.Q923X) запідозрено дифузну форму, і в одного пацієнта генетичну природу ВГІ встановити не вдалося. 6 пацієнтів були скеровані для подальшого дообстеження й лікування до центру гіперінсулінізму в м. Оденсе, Данія (батьки дитини з мутацією ABCC8, p.Q444H/p.Q923X відмовилися від подальшого обстеження). За результатами ПЕТ з 18-F-допою в чотирьох пацієнтів підтверджено фокальну форму ВГІ, у двох — дифузну форму (у дитини без генетичної мутації та в дитини з ABCC8 p.R1251/p.Y1287) (рис. 1, 2).
/169/169.jpg)
У зв’язку із недостатньою ефективністю консервативної терапії чотирьом пацієнтам з фокальною формою було виконано хірургічне втручання — інвагінаційну панкреатогастростомію після резекції фокуса в голівці підшлункової залози, дитині з атиповою формою проведено панкреатичну резекцію 80 %, дитині з дифузною формою — резекцію 90 % підшлункової залози.
Гістологічне дослідження підтвердило фокальну форму ВГІ у п’яти пацієнтів, дифузну форму підтверджено в одного пацієнта. У дитини без генетичної мутації верифіковано атипову форму ВГІ.
Слід зауважити, що в однієї дитини (з мутацією ABCC8, p.Q444H) через 6 міс. після операції розвинулася післяопераційна кіста, в однієї дитини (з мутацією KCNJ11, p.F333S) спостерігалася транзиторна гіперглікемія натще, що через 2 міс. минула самостійно.
У дитини з дифузною (атиповою) формою ВГІ через 1 місяць після операції з’явилися скарги на часті випорожнення з неприємним запахом, при обстеженні виявлено низький рівень панкреатичної еластази, розпочато лікування ферментами (креон 10 000 ОД 2 рази на добу) з покращенням стану.
Слід зазначити, що у жодного пацієнта надалі не спостерігали стійких порушень вуглеводного обміну.
Двоє дітей (з мутаціями ABCC8, p.Q444H та з c.4415–13G>A) до і після операції мали тяжку затримку психомоторного розвитку, часті судоми (на тлі постійного прийому протисудомних препаратів), у зв’язку із неефективністю останніх їм було проведено пульс-терапію кортикостероїдами з досягненням стійкого ефекту (зникненням судом). Найбільш тяжке ураження центральної нервової системи (у вигляді сліпоти, неможливості розмовляти, тримати голову, сидіти або ходити) спостерігали у дитини з ABCC8, p.Q444H. На жаль, оперативне втручання, що було проведено у віці 9 міс., було надто пізнім і надалі не призвело до покращення психомоторного розвитку.
Отже, ВГІ є потенційно небезпечним захворюванням, оскільки тяжкі, некеровані гіпоглікемії можуть призвести як до смерті дитини, так і до тяжкого, необоротного ураження головного мозку. Складність досягнення нормоглікемії завдяки медикаментозній терапії диктує необхідність своєчасного оперативного лікування, що має проводитися в спеціалізованих центрах досвідченою бригадою хірургів. Незважаючи на прорив в дослідженні етіології та патогенезу ВГІ, у 50 % випадків молекулярно-генетичний діагноз залишається неуточненим, що потребує подальших досліджень у цій галузі.
Висновки
1. Усім дітям з гіпоглікеміями, підвищеним (або нормальним) рівнем С-пептиду та інсуліну необхідно проводити генетичну діагностику та ПЕТ з 18-F-допою для уточнення типу ВГІ та вибору лікувальної тактики.
2. Необхідний подальший розвиток генетичної діагностики для пошуку нових генів ВГІ.
3. Оперативне лікування може бути терапією вибору в дітей з ВГІ, особливо при його фокальній формі.
1. James C., Kapoor R.R., Ismail D. et al. The genetic basis of congenital hyperinsulinism // J. Med. Genet. — 2009. — 46. — 289–299.
2. Otonkoski T., Ammala C., Huopio H. et al. A point mutation inactivating the sulfonylurea receptor causes the severe form of persistent hyperinsulinemic hypoglycemia of infancy in Finland // Diabetes. — 1999. — 48. —408–415.
3. De Leon D.D., Stanley C.A. Mechanisms of disease: advances in diagnosis and treatment of hyperinsulinism in neonates // Nat. Clin. Pract. Endocrinol. Metab. — 2007. — 3. — 57–68.
4. McQuarrie I. Idiopathic spontaneously occurring hypoglycemia in infants: clinical significance of problem and treatment // AMA Am. J. Dis. Child. — 1954. — 87. — 399–428.
5. Rahier J., Guiot Y., Sempoux C. Persistent hyperinsulinaemic hypoglycaemia of infancy: a heterogeneous syndrome unrelated to nesidioblastosis // Arch. Dis. Child Fetal. Neonatal. — 2000. — 82. — F108–F112.
6. Wolfsdorf J.I., Weinstein D.A. Hypoglycemia in Children // Pediatric Endocrinology. — 5th Ed. — NY: Blackwell Munksgaard, 2007. — 1. — 291–327.
7. Kapoor R.R., Flanagan S.E., James C. et al. Hyperinsulinaemic hypoglycaemia // Arch. Dis. Child. — 2009. — 94. — 450–457.
8. Kapoor R.R., James C., Hussain K. Advances in the diagnosis and management of hyperinsulinemic hypoglycemia // Nat., Clin. Pract. Endocrinol. Metab. — 2009. — 5, 2. — 101–112.
9. Thomas P., Ye Y., Lightner E. Mutation of the pancreatic islet inward rectifi er Kir6.2 also leads to familial persistent hyperinsulinemic hypoglycemia of infancy // Hum. Mol. Genet. — 1996. — 5. — 1809–1812.
10. Thomas P.M., Cote G.J., Wohllk N. et al. Mutations in the sulfonylurea receptor gene in familial persistent hyperinsulinemic hypoglycemia of infancy // Science. — 1995. — 268. — 426–429.
11. Nestorowicz A., Inagaki N., Gonoi T. et al. A nonsense mutation in the inward rectifi er potassium channel gene, Kir6.2, is associated with familial hyperinsulinism // Diabetes. — 1997. — 46. — 1743–1748.
12. Dunne M.J., Kane C., Shepherd R.M. et al. Familial persistent hyperinsulinemic hypoglycemia of infancy and mutations in the sulfonylurea receptor // N. Engl. J. Med. — 1997. — 336. — 703–706.
13. Christesen H.B., Brusgaard K., Beck Nielsen H., Brock Jacobsen B. Non-insulinoma persistent hyperinsulinaemic hypoglycaemia caused by an activating glucokinase mutation: hypoglycaemia unawareness and attacks // Clin. Endocrinol. (Oxford). — 2008. — 68. — 1011.
14. Hussain K., Blankenstein O., De Lonlay P., Christesen H.T. Hyperinsulinaemic hypoglycaemia: biochemical basis and the importance of maintaining normoglycaemia during management // Arch. Dis. Child. — 2007. — 92. — 568–570.
15. Suchi М., MacMullen С.М., Thornton P.S., Adzick N.S. Molecular and immunohistochemical analyses of the focal form of congenital hyperinsulinism // Modern Pathology. — 2006. — 19. — 122–129.
16. Lonlay P., Simon-Carre A., Ribeiro М.-J. et al. Congenital Hyperinsulinism: Pancreatic [18F] Fluoro LDihydroxyphenylalanine (DOPA) Positron Emission Tomography and Immunohistochemistry Study of DOPA Decarboxylase and Insulin // J. Clin. Endocrinol. Metab. — 2006 Mar. — 91 (3). — 933–40.
17. de Lonlay P., Fournet J.C., Touati G. et al. Heterogeneity of persistent hyperinsulinaemic hypoglycaemia. A series of 175 cases // Eur. J. Pediatr. — 2002. — 161. — 37–48.
18. Cherubini V., Bagalini L.S., Ianilli A. et al. Rapid genetic analysis, imaging with 18F-DOPA-PET/CT scan and laparoscopic surgery in congenital hyperinsulinism // J. Pediatr. Endocrinol. Metab. — 2010. — 23 (1–2). — 171–177.