Статья опубликована на с. 153-159
Расстройства аутистического спектра (РАС) — одна из наиболее загадочных проблем современной нейропедиатрии. В последнее десятилетие данная проблема не перестает привлекать внимание специалистов разных профилей. Тот факт, что детские неврологи в нашей стране проявляют высокий интерес к РАС, нельзя не расценивать как позитивный. Находясь ранее исключительно в сфере компетенции детских психиатров, аутизм имел стойкую репутацию психического заболевания, что в немалой степени способствовало стигматизации пациентов с этим диагнозом, а также порождало миф о его полной неизлечимости.
На сегодняшний день РАС определяются как гетерогенная группа нарушений развития нервной системы, которые имеют разнообразную этиологию, но характеризуются общими симптомами, связанными с нарушениями социального взаимодействия, коммуникации и поведения (в частности, стереотипность и ограниченность действий или интересов). Таким образом, РАС рассматриваются как поведенческое нарушение, однако в основе его лежит взаимодействие множества генетических факторов и окружающей среды, приводящих к нарушению развития нервной системы [1].
Распространенность РАС в мире с каждым годом стремительно увеличивается, что привело к появлению выражения «эпидемия РАС». По последним данным Центра по контролю и профилактике заболеваний (Centers for Disease Control and Prevention, USA), распространенность РАС среди детей составляет 1 : 68, в т.ч. среди мальчиков — 1 : 42 (14,7 на 1000 детей в возрасте 8 лет), что на 30 % больше, чем в 2012 году (1 : 88) [2]. В Южной Корее распространенность РАС среди детей в возрасте от 7 до 14 лет оценивается как соотношение 1 : 38 [3].
В клинике детской психоневрологии ГУ «Институт педиатрии, акушерства и гинекологии НАМН Украины» мы наблюдаем пациентов с РАС, в том числе сочетающимися с эпилептическими приступами. Ранее мы уже обращались к проблеме взаимосвязи РАС и эпилептических энцефалопатий, выразив мнение, что в большинстве случаев у детей раннего возраста именно резистентные эпилептические приступы либо персистирующая эпилептиформная активность приводят к формированию РАС [4].
На сегодняшний день многие исследования подтверждают наличие взаимосвязи между эпилептическими приступами, эпилептиформными изменениями на электроэнцефалограмме (ЭЭГ) и расстройствами аутистического спектра, однако ее механизмы остаются до конца не понятными. Высокая распространенность эпилептических приступов среди детей с РАС, как и значительный уровень заболеваемости РАС среди пациентов с эпилептическими приступами, указывает на возможное существование общих причин, лежащих в основе этих двух патологических процессов.
По данным метаанализа исследований, проводившихся с 1963 по 2006 г., средняя частота развития эпилептических приступов составила 21,5 % у лиц с РАС при наличии низкого IQ и 8 % у пациентов с сохранным интеллектом [5]. Хотя высокая частота эпилептических приступов среди больных с РАС характерна преимущественно для детей со сниженным интеллектом (часто имеющих выраженные структурные изменения головного мозга или генетическую патологию), у детей с РАС и сохранным интеллектом риск развития эпилептических приступов в 8 раз выше, чем в популяции. Условно можно выделить две группы детей с наличием эпилептических приступов и РАС: 1) пациенты с эпилептическими приступами или персистирующей эпилептиформной активностью на ЭЭГ, у которых развивается регресс коммуникативных и социальных навыков по аутистическому типу; 2) пациенты с РАС, у которых со временем развиваются эпилептические приступы.
По мнению зарубежных исследователей, не менее 30 % детей с эпилепсией имеют РАС [6]. Проведено также два крупных проспективных исследования, посвященных изучению распространенности РАС среди детей с эпилепсией, которые выявили ее на уровне примерно 4–5 % [7, 8]. Наиболее высокий риск развития РАС при эпилепсии имеют дети с умственной отсталостью. Однако даже при отсутствии умственной отсталости риск РАС при эпилепсии значительно выше, чем в общей популяции.
Анализ показателей частоты сочетания (коморбидности) РАС и эпилепсии позволил создать гипотезу общности патофизиологических механизмов двух патологий. Исследователями в области нейрофизиологии высказано предположение, что генез РАС и эпилепсии может быть объяснен с точки зрения нарушений синаптической пластичности, которые приводит к дисбалансу возбуждения и торможения в развивающемся мозге [9].
Синаптическая пластичность — это возможность изменения силы синапса (величины изменения трансмембранного потенциала) в ответ на изменение его активности [10]. Это уникальное свойство мозга млекопитающих лежит в основе развития сложных форм поведения, основанных на усвоенном опыте, мышлении, памяти, эмоциях. Кроме того, синаптическая пластичность играет важную роль в раннем развитии нервной системы, а нарушение механизмов пластичности все чаще связывают с возникновением различных нервно-психических расстройств.
Патологические изменения в мозге, приводящие к развитию РАС и эпилепсии, характерны для генетических заболеваний, таких как туберозный склероз, синдромы Х-ломкой хромосомы и Ретта, а также при возникающих de novo редких вариациях числа копий генов (copy number variants (CNVs), например, мутации нейролигина/нейрексина или мутации SHANK3, а также интернейропатиях, вызванных мутациями гена ARX (Aristaless related homeobox) или нейропилина-2 (NRP2), и других мутациях [8].
Вариация числа копий генов (CNV) — вид генетического полиморфизма, к которому относят различия индивидуальных геномов по числу копий хромосомных сегментов размером от 1 тыс. до нескольких миллионов пар оснований. Впервые сведения о новом типе вариаций в геноме человека — CNVs — опубликованы R. Redon и соавт. в 2006 году в журнале «Nature». На сегодняшний день известно, что CNV связаны с 17 заболеваниями, в том числе и эндогенными психическими [11]. CNV возникают, как правило, в результате мутаций de novo, таких как делеции, дупликации, инверсии или транслокации.
Роль CNV в этиологии РАС была установлена в 2007–2008 гг. в рамках крупного исследования Autism Genome Project, в процессе которого были проанализированы геномы 2611 семей пациентов с РАС.
Одной из таких CNV, приводящих к развитию РАС и эпилепсии, является 2p16 — делеция в гене 2-й хромосомы, который кодирует белок нейрексин (NRXN1). NRXN1 взаимодействует с нейролигином — белком клеточной адгезии, обеспечивающим коммуникацию нейронов. Взаимодействие нейрексина и нейролигина позволяет формировать нейрональные пути в головном мозге. Кроме того, комплекс нейрексин-нейролигин важен для функционирования глутаматных синапсов, аномальная работа которых рассматривается как одна из причин развития РАС [10]. К подобным изменениям в ЦНС также приводит делеция 22q13.3, при которой отсутствует ген, кодирующий протеин SHANK3, который играет важную роль в разветвлении дендритов и формировании синапсов [12].
Мутация гена ARX, кодирующего Х-сцепленный протеин aristaless-related homeobox, ответственный за транскрипцию ДНК в ЦНС, приводит к сокращению количества интернейронов коры полушарий и гиппокампа, что вызывает тяжелые эпилептические приступы с ранним дебютом (чаще в виде инфантильных спазмов) и РАС [13].
Нейропилин-2 (NRP) представляет собой рецептор для аксонального медиатора семафорина-3F (SEMA3F). Полиморфизм гена NRP2 может приводить к развитию эпилептических приступов и РАС. Изменения в мозге при этой мутации включают изменения количества интернейронов гиппокампа, нарушение функционирования парвальбумина и нейропептида Y и сокращение длины дендритов пирамидальных нейронов СА1 [14, 15].
По мнению американского специалиста в сфере РАС и эпилепсии Amy Brooks-Kayal, «…независимо от и в дополнение к генетически обусловленным нарушениям синаптической пластичности в развивающемся мозге могут возникать изменения, обусловленные эпилептогенезом, которые могут приводить к нарушениям синаптической пластичности и способствовать развитию РАС». Происходящие в мозге эпилептогенез и эпилептические приступы нарушают нормальное течение процессов развития в мозге, включая синаптический прунинг, истончение дендритов и аксонов и созревание рецепторов ионных каналов [8].
Одним из негативных эффектов эпилептогенеза на мозг является нарушение работы нейротрансмиттерных систем. В частности, под влиянием рано возникших эпилептических приступов происходит изменение действия гамма-аминомасляной кислоты (ГАМК) на нейроны гиппокампа СА1 с деполяризующего (ингибирующего) на гиперполяризирующее. Деполяризующее действие ГАМК критически важно для множества нормальных процессов развития, включая пролиферацию нейронов, миграцию, таргетинг и синаптогенез. Возможно, что изменение возбуждающих и тормозных процессов в синапсах также может способствовать развитию эпилептических приступов и РАС в созревающем мозге.
Таким образом, во многих случаях именно эпилептическая энцефалопатия (эпилептиформная активность и эпилептические приступы), развивающаяся в генетически скомпрометированном мозге, является фактором, который приводит к тяжелым когнитивным и поведенческим нарушениям и формированию РАС [16–18, 30].
Концепция общего патогенеза, лежащего в основе эпилепсии и РАС, имеет важное значение для разработки принципов лечения, однако до конца механизмы нарушений работы мозга при этих состояниях не ясны.
Примером эпилептической энцефалопатии, приведшей к развитию РАС у ребенка с генетической патологией, мы хотели бы поделиться в данной работе.
Синдром XXYY был впервые описан Sylfest Muldal и Charles H. Ockey в 1964 году в Манчестере (Великобритания) на примере 15-летнего умственно отсталого мальчика с кариотипом 48, XXYY. Пациент имел фенотипические признаки синдрома Кляйнфельтера, однако вместо характерного для этого синдрома хромосомного набора 47, XXY исследование кариотипа выявило увеличение количества хромосом — 48, XXYY. Поэтому синдром XXYY первоначально считался вариантом синдрома Кляйнфельтера [19]. Соавтор первого описания синдрома XXYY, Sylfest Muldal, признан одним из пионеров в области генетических исследований в Великобритании. Родившись в Норвегии, он в 1946 г. переехал в Британию, где начал научный путь в John Innes Institute (г. Уимблдон) под руководством профессора Cyril Dean Darlington, патриарха британской генетики, открывшего, в частности, механизм хромосомного кроссинговера (процесс обмена участками гомологичных хромосом во время конъюгации в профазе I мейоза). Позже Sylfest Muldal переехал в Манчестер, где ему было предложено создать лабораторию цитологии и генетики для исследования хромосомных аномалий с целью диагностики специфических типов рака. Он также воспитал целую плеяду генетиков и клиницистов, наиболее известный из которых — онколог Sir David Philip Lane, первооткрыватель протеина — супрессора роста опухолей р53.
Частота возникновения синдрома XXYY составляет примерно 1 на 18 000–50 000 лиц мужского пола и занимает примерно 0,08–0,33 % среди мальчиков с умственной отсталостью [20, 21]. На сегодняшний день в научной литературе описано около 100 случаев синдрома XXYY, который ранее считался вариантом синдрома Кляйнфельтера (47, XXY) благодаря схожему фенотипу и наличию эндокринологических расстройств. Характерные для этих синдромов признаки включают высокий рост, микроорхидизм, гипергонадотропный гипогонадизм [22]. Хотя первые сообщения о синдроме XXYY не показывали существенных различий между ним и синдромом Кляйнфельтера [23], сегодня общепризнанным фактом является существование синдрома 48, XXYY как отдельной нозологических единицы, т.к. болезнь характеризуется собственными особенностями нервной и эндокринной систем, а также поведенческими расстройствами [24].
Дополнительные хромосомы при кариотипе XXYY почти всегда происходят от сперматозоидов с тремя половыми хромосомами (одна Х-хромосома и две Y-хромосомы). Это случается из-за нерасхождения хромосом во время 1-го и 2-го делений мейоза. Кариотип 48, XXYY в некоторых случаях возникает в результате нерасхождения хромосом при митозе зиготы с нормальным кариотипом 46, XY [25].
Пациенты с синдромом XXYY имеют фенотип, напоминающий синдром Кляйнфельтера: высокий рост, гипогонадизм, гинекомастия. Эндокринные нарушения становятся выраженными в пубертатном периоде, когда возникает задержка развития вторичных половых признаков: снижение оволосения на лице и теле, гинекомастия, слабое развитие мускулатуры. В некоторых случаях описаны гипертелоризм, варгусная деформация локтевого сустава, плоскостопие, косолапость, клинодактилия и сколиоз. Примерно у 71 % пациентов наблюдается тремор, который усугубляется с возрастом. У многих пациентов также выявляется неврологическая симтоматика: эпилептические приступы, мышечная гипотония, тики.
На МРТ часто выявляют структурные нарушения, такие как агенезия мозолистого тела или атрофия лобно-височных долей мозга [26]. Примерно у 50 % на МРТ выявляются неспецифические изменения в виде гипер-интенсивного сигнала от белого вещества на T2/Flair, мелкие (1–2 мм) или более крупные (4–5 мм) локальные очаги в белом веществе. Также около 8 % пациентов имеют врожденные пороки сердца, такие как тетрада Фалло, стеноз устья легочной артерии, пролапс митрального клапана или дефекты перегородок. Среди сопутствующих заболеваний и симптомов описаны косоглазие, паховая грыжа, крипторхизм, астма, рецидивирующие респираторные инфекции, нарушение роста зубов. У взрослых нередки случаи тромбоза глубоких вен нижних конечностей. Упоминается также повышенный уровень смертности от неходжкинской лимфомы.
Практически у всех детей с синдромом XXYY отмечается задержка психического и речевого развития, которая затем переходит в расстройство аутистического спектра или умственную отсталость. Характерны также нарушения поведения в виде синдрома гиперактивности с дефицитом внимания, импульсивность, агрессия, нарушения настроения. Также до 3 лет задержано развитие статокинетических функций [26, 27].
Предполагается, что высокий рост пациентов с синдромом XXYY связан с повышенной экспрессией гена SHOX, что способствует избыточной активности ростовых пластинок трубчатых костей и ускоренному созреванию костной ткани [28].
В качестве иллюстрации приводим клиническое наблюдение сочетания эпилептической энцефалопатии и РАС у ребенка с диагностированным синдромом XXYY.
Мальчик Р. впервые поступил в клинику детской психоневрологии ГУ «Институт педиатрии, акушерства и гинекологии НАМН Украины» весной 2012 г. в возрасте 2 лет. При поступлении родителями предъявлялись жалобы на наличие у ребенка эпилептических приступов в виде тонического напряжения и миоклоний конечностей и туловища длительностью 2–3 минуты, которые возникали только в ночное время. Приступы сопровождались вокализациями (звуками, напоминающими рычание). Также у ребенка отмечалась задержка психоэмоционального развития: практически отсутствовал контакт с окружающими, отсутствовала экспрессивная речь, адекватная реакция на обращенную речь, не выполнялись просьбы родителей.
Из анамнеза было известно, что ребенок родился от 2-й беременности, которая протекала без патологии. От 1-й беременности у матери есть здоровый мальчик 5 лет. Мать до первой беременности перенесла черепно-мозговую травму, вследствие чего как первые, так и вторые роды проводились путем планового кесарева сечения. Ребенок родился на 40-й неделе беременности с массой 2500 г (1-й ребенок — 2270 г), длиной тела 47 см. После рождения отмечался цианоз кожных покровов, закричал после санации дыхательных путей. По шкале Апгар получил 7 баллов. Отмечалась незначительная желтуха 2–3 нед., билирубин был максимально 108 ммоль/л. Выписан из роддома на 9-й день. Голову начал держать с 3 мес., сидеть — с 5 мес., ходить — в 1 г. 5 мес. Вовремя начал следить за игрушкой, эмоционально реагировать на окружающих, улыбаться.
В 9 мес. на фоне полного здоровья мама заметила у ребенка приступы в виде сгибания и приведения конечностей, сгибания туловища. Приступы отмечались 2–3 раза в день, сериями (максимально 10 приступов в серии). Во время приступов ребенок застывал, смотрел в одну точку. По месту жительства ребенку был назначен депакин (20 мг/кг), на фоне приема которого дневные приступы исчезли, однако появились в ночное время, при этом ребенок просыпался, плакал, также при приступах отмечались вокализации в виде звуков, напоминающих рычание. Кроме генерализованных приступов в ночное время также периодически отмечались миоклонии мышц туловища и конечностей. Семейный анамнез со слов матери не отягощен. Также с 8 мес. у ребенка несколько раз в год возникали эпизоды рвоты, которые расценивались педиатрами как проявления ацетонемического синдрома.
При осмотре ребенка обращало на себя внимание наличие нарушения психического развития по типу расстройства аутистического спектра. Ребенок практически не реагировал на обращенную речь, не выполнял инструкции родителей. Характер поведения был гиперактивным, бесцельным, с частыми стереотипными движениями рук. Периодически реагировал на собственное имя. Сюжетная игра отсутствовала, предпочитал игрушкам неигровые предметы (например, посуду). Окружность головы 47,4 см (3-й перцентиль). Рост 95 см (50-й перцентиль). Вес 12,5 кг. Зубы, язык, небо не изменены. Гипертелоризм. Выражены проявления дисплазии соединительной ткани (гипермобильный суставной синдром, пролапс митрального клапана, плоскостопие, крипторхизм). Кожные покровы сухие. Сидит самостоятельно, берет предметы в руки. Походка неуверенная, с элементами шаткости. Патология черепных нервов отсутствует. Мышечный тонус несколько снижен. Сухожильные рефлексы с конечностей живые. D = S. Брюшные рефлексы положительные.
При проведении МРТ головного мозга выявлены признаки гипоплазии головного мозга в лобных областях.
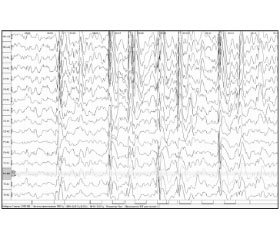
На ЭЭГ выявлена продолженная, с проявлениями билатеральной синхронизации эпилептиформная активность в виде генерализованных разрядов комплексов острая-медленная волна, пик — медленная волна частотой 1,5–2,0 Гц в сочетании с высокоамплитудными медленными волнами, с амплитудным преобладанием в заднелобных, центральновисочных отведениях слева (рис. 1, 2). Обследование проводилось на 16-канальном электроэнцефалографе «Нейрон-спектр».
С учетом наличия у ребенка сочетания резистентных к терапии эпилептических приступов и расстройства аутистического спектра было сделано предположение о нейрометаболической этиологии заболевания. Генетиком был назначен анализ на определение уровня молочной и пировиноградной кислот в крови.
При проведении лабораторного исследования в сыворотке крови отмечалось увеличение содержания молочной (2,07 ммоль/л (N 1,0–1,7 ммоль/л)) и пировиноградной кислоты (0,22 ммоль/л (N 0,09–0,11 ммоль/л)). На фоне стандартного глюкозотолерантного теста уровень лактата повысился до 3,86 ммоль/л через 2 часа. Уровень пирувата через 2 ч достиг 0,28 ммоль/л. Содержание аминокислот в плазме крови находилось в пределах нормы.
Результаты остальных лабораторно-инструментальных методов исследования (общий и биохимический анализ крови, общий анализ мочи, ЭКГ, ЭхоКГ, УЗИ органов брюшной полости, осмотр окулиста) патологических изменений не показали.
Консультации психиатра и медицинского психолога подтвердили наличие у ребенка расстройств аутистического спектра в соответствии с критериями DSM-V. Так как выраженность проявлений когнитивных нарушений усугублялась частыми эпилептическими приступами, состояние ребенка было расценено как эпилептическая энцефалопатия.
С учетом наличия у ребенка признаков митохондриальной недостаточности (лактат-ацидоз) было принято решение провести замену депакина, как препарата, негативно влияющего на энергетический обмен, на леветирацетам (кеппру) в суточной дозе 40 мг/кг. Кроме того, проводилась коррекция лактат-ацидоза и когнитивных нарушений с помощью препаратов аддитивного (нейрометаболического) действия: цитиколина, L-карнитина, глицина, янтарной кислоты, магния сульфата, витами-на В6. Ребенок выписан с диагнозом: ранняя эпилептическая нейрометаболическая энцефалопатия с выраженными психоэмоциональными нарушениями в виде расстройства аутистического спектра.
С целью потенцирования антиконвульсивного эффекта терапии и дальнейшей терапии энцефалопатии к лечению был добавлен метилпреднизолон в дозе 1 мг/кг/сут на срок 2 месяца, с постепенным снижением дозы. Продолжалось также лечение, направленное на коррекцию митохондриальной дисфункции: L-карнитин, коэнзим-Q10, витамины группы В, янтарная кислота. При осмотре через 2 мес. выявлена позитивная динамика состояния в виде отсутствия эпилептических приступов, улучшения реакции ребенка на окружающих, более упорядоченного поведения.
Родители по собственной инициативе прошли обследование в Институте генетики человека им. Данека Гертнера (Израиль), в ходе которого были исключены синдром Х-ломкой хромосомы, а также патология гена ARX. Также было предположено наличие у ребенка кариотипа 47, ХХУ, для уточнения рекомендовано определение кариотипа в Украине.
Ремиссия эпилептических приступов сохранялась около 1 года, после чего возобновились приступы в виде кивков головы, расширения зрачков, отсутствия сознания в течение нескольких секунд. Ребенок вторично поступил на обследование и лечение в клинику детской психоневрологии ГУ «Институт педиатрии, акушерства и гинекологии НАМН Украины». Проведена коррекция антиконвульсивной терапии: добавлен к лечению этосукцимид (20 мг/кг/сут) в сочетании с препаратами нейрометаболического действия (цитиколин, цитофлавин, глицин, витамины группы В). В то же время проведен анализ на определение кариотипа в лаборатории клиники «Исида» (г. Киев) методом GTG-бэндинга, в результате которого получен результат о наличии у ребенка хромосомного набора 48, XXYY. После получения результатов анализа на кариотип были обследованы родители ребенка, однако патологических отклонений хромосомных наборов у них выявлено не было. Таким образом, на основании данных анамнеза, клинического осмотра и результатов лабораторно-инструментальных методов обследования нами был установлен диагноз: синдром XXYY как пример сочетания метаболической эпилептической энцефалопатии и расстройства аутистического спектра с генетической детерминацией.
Приведенный выше случай наглядно иллюстрирует всю сложность диагностического поиска при расстройствах аутистического спектра у детей. У данного ребенка мы наблюдали сочетание тяжелых фармакорезистентных эпилептических приступов с РАС, и только использование генетических исследований позволило нам выявить этиологию клинических симптомов — хромосомный синдром. Публикуя данное наблюдение, мы хотели бы сделать акцент на важности обследования неврологом детей с РАС. Отсутствие тщательного обследования уводит нас от поиска этиологии заболевания у конкретного пациента, а значит, во многих случаях не назначается адекватное лечение. Мы считаем, что актуальное представление о РАС как о результате нарушения развития мозга делает недопустимым ситуацию, когда обследование ребенка с РАС заканчивается на консультации психиатра. Особое внимание следует уделить развитию в нашей стране материально-технической базы для обследования детей с РАС: генетических и биохимических методов обследования, ЭЭГ, магнитно-резонансной томографии.
Отдельно следует остановиться на проблеме лечения детей с РАС. Мы считаем, что подбор терапии в каждом случае должен осуществляться индивидуально, с учетом результатов клинико-инструментального обследования. В приведенном выше клиническом примере показано, как биохимические обследования (определение уровня лактата и пирувата) позволили выявить митохондриальную недостаточность у ребенка и, как результат, заподозрить генетическое заболевание. Ранее в нашей работе, посвященной тяжелому митохондриальному заболеванию — синдрому Лея, мы уже освещали принципы коррекции митохондриальной недостаточности у детей. Назначают препараты, стимулирующие синтез АТФ в митохондриях и снижающие уровень лактата: коэнзим Q10 (4 мг/кг/сут), витамин Е (200–400 мг/сут, фолиевая кислота (0,5–2,5 мг/кг/с), L-аргинин (500 мг/кг/сут, внутривенно капельно), тиамин и рибофлавин (по 50 мг/сут), L-карнитин (30–50 мг/кг/сут) [29]. Важен также индивидуальный подбор антиконвульсивной, а при необходимости — психотропной терапии. Следует помнить, что назначение психотропных препаратов, находящееся в компетенции детских психиатров, возможно лишь с 5 лет. До этого возраста из медикаментозных препаратов в нашем арсенале остаются только антиконвульсанты и препараты нейрометаболического и нейропротекторного действия.
Список литературы
1. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. — 4th Edition, Text Revision (DSM-IV-TR). — Washington: American Psychiatric Publishing, 2000.
2. CDC estimates 1 in 68 children has been identified with autism spectrum disorder. Press Release CDC Division of News & Electronic Media. Centers for Disease Control and Prevention. March 27, 2014/Available on http: // www.cdc.gov/media/releases/2014/p0327-autism-spectrum-disorder.html
3. Kim Y.S., Leventhal B.L., Koh Y.J. et al. Prevalence of autism spectrum disorders in a total population sample // Am. J. Psychiatry. — 2011. — 168 (9). — 904-12. doi: 10.1176/appi.ajp.2011.10101532. Epub 2011 May 9.
4. Кирилова Л.Г, Мірошников О.О. Погляд невролога на проблему розладів аутистичного спектру у дітей // Соціальна педіатрія та реабілітологія. — 2013. — № 2 (5). — С. 66-72.
5. Amiet C., Gourfinkel-An I., Bouzamondo A. et al. Epilepsyin autism is associated with intellectual disability and gender: evidence from a meta-analysis // Biol. Psychiatry. — 2008. — 64. — 577-582.
6. Tuchman R., Moshé S.L., Rapin I. Review Convulsing toward the pathophysiology of autism // Brain Dev. — 2009. — 31 (2). — 95-103.
7. Geerts A., Brouwer O., van Donselaar C. et al. Health perception and socioeconomic status following childhood-onset epilepsy: the Dutch study of epilepsy in childhood // Epilepsia. — 2011. — 52. — 2192-2202.
8. Berg A.T., Plioplys S., Tuchman R. Risk and correlates of autism spectrum disorder in children with epilepsy: a community-based study // J. Child Neurol. — 2011. — 26. — 540-547.
9. Amy Brooks-Kayal Epilepsy and autism spectrum disorders: Are there common developmental mechanisms? // Brain & Development — 2010. — 32. — Р. 731-738.
10. Citri A., Malenka R.C. Synaptic plasticity: multiple forms, functions, and mechanisms // Neuropsychopharmacology. — 2008. — 33 (1). — 18-41. Epub 2007 Aug 29.
11. Голимбет В.Е., Корень Е.В. Вариации числа копий в геноме — новая страница в генетических исследованиях в области психиатрии: международный проект PsychCNVs // Журнал неврологии и психиатрии им. С.С. Корсакова. — 2010. — № 1. — С. 107-109.
12. Евтушенко С.К., Морозова Т.М. и др. Идентификация мутаций у ребенка с митохондриальной энцефалопатией // Международный неврологический журнал. — 2010. — № 3 (32). — С. 64-70.
13. Szatmari P., Paterson A.D., Zwaigenbaum L., Roberts W., Brian J. et. al. Autism Genome Project Consortium. Mapping autism risk loci using genetic linkage and chromosomal rearrangements // Nat. Genet. — 2007. — 39 (3). — 319-28.
14. Boeckers T.M. et al. ProSAP/Shank proteins — a family of higher order organizing molecules of the postsynaptic density with an emerging role in human neurological disease // J. Neurochem. — 2002. — 81 (5). — 903-10.
15. Marsh E., Fulp C., Gomez E. et al. Targeted loss of Arx results in a developmental epilepsy mouse model and recapitulates the human phenotype in heterozygous females // Brain. — 2009. — 132 (Pt 6). — 1563-76.
16. Wu S., Yue W., Jia M. et аl. Association of the neuropilin-2 (NRP2) gene polymorphisms with autism in Chinese Han population // Am. J. Med. Genet. B. Neuropsychiatr. Genet. — 2007. — 144B (4). — 492-5.
17. Gant J.C., Thibault O., Blalock E.M. et al. Decreased number of interneurons and increased seizures in neuropilin 2 deficient mice: implications for autism and epilepsy // Epilepsia. — 2009. — 50 (4). — 629-45.
18. Jensen F.E. Epilepsy as a spectrum disorder: implications from novel clinical and basic neuroscience // Epilepsia. — 2011. — 52 (suppl. 1). — 1-6.
19. Galanopoulou A.S., Moshe S.L. The epileptic hypothesis: developmentally related arguments based on animal models // Epilepsia. — 2009. — 50 (suppl. 7). — 37-42.
20. Rakhade S.N., Jensen F.E. Epileptogenesis in the immature brain: emerging mechanisms // Nat. Rev. Neurol. — 2009. — 5. — 380-391.
21. Muldal S., Ockey C.H. Double male: New chromosome constitution in Klinefelter's syndrome // Lancet. — 1960. — 2. — 492-493.
22. Maclean N., Mitchell J.M. A survey of sex-chromosome abnormalities among 4,514 mental defectives // Lancet. — 1962. — 1. — 293-296.
23. Sorensen K., Nielsen J., Jacobsen P., Rolle T. The 48, XXYY syndrome // Ment. Defic. Res. — 1978. — 22. — 197-205.
24. Евтушенко С.К. Неординарные (раритетные) синдромы и заболевания нервной системы у детей и взрослых // Мат-лы Междунар. науч.-практ. конф. / Под ред. проф. С.К. Евтушенко. — Донецк; Святогорск. — 2003. — 398 с.
25. Townes P.L., Ziegler N.A., Scheiner A.P. An XXYY variant of the Klinefelter syndrome in a prepubertal body // J. Pediatr. — 1965. — 67. — 410-414.
26. Tartaglia N., Davis S., Hench A., Nimishakavi S., Beauregard R., Reynolds A., Fenton L., Albrecht L., Ross J., Visootsak J., Hansen R., Hagerman R. A new look at XXYY syndrome: medical and psychological features // Am. J. Med. Genet A. — 2008. — 15. — 146A (12). — 1509-22. doi: 10.1002/ajmg.a.32366.
27. Genetic Home Reference. XXYY Syndrome. — 2010 Jan, [cited 2011 Jun 16].
28. Munns C.J., Haase H.R., Crowther L.M., Hayes M.T., Blaschke R., Rappold G., Glass I.A., Batch J.A. Expression of SHOX in human fetal and childhood growth plate // J. Clin. Endocrinol. Metab. — 2004. — 89 (8). — 4130-5.
29. Раритетне захворювання у нейропедіатрії — синдром Лея [Текст] / Л.Г. Кирилова [та ін.] // Педіатрія, акушерство та гінекологія. — 2013. — Т. 75, № 1. — С. 46-51.
30. Евтушенко С.К. Резидуальная энцефалопатия, или незнание, возведенное в диагноз // Международный неврологический журнал. — 2010. — № 7 (37). — С. 105-110.

/157/157.jpg)