Статья опубликована на с. 60-72
Болезнь Паркинсона — хроническое прогрессирующее заболевание головного мозга с дегенерацией нигростриарных нейронов и нарушением функции базальных ганглиев с клинической симптоматикой экстрапирамидной патологии.
Распространенность паркинсонизма: 60–140 случаев на 100 000 населения. В Украине 140 случаев на 100 000 населения (Карабань И.Н., 2010; Труфанов Е.В., 2012).
Основу экстрапирамидной системы (стриатума) составляют базальные ганглии.
К базальным ганглиям принято относить 5 основных ядер:
— скорлупу — putamen,
— хвостатое ядро — n. caudatus,
— бледный шар — globus pallidus,
— черную субстанцию — substantia nigra,
— субталамическое ядро — n. subthalamicus.
Базальные ганглии и различные зоны коры представляют собой структурно и функционально обособленные друг от друга относительно замкнутые нейронные круги (петли).
На состояние сегментарного аппарата спинного мозга оказывают влияние не только кортикоспинальные, но и стволово-спинальные пути. Стволово-спинальные системы подразделяют на медиальную (вентромедиальную) и латеральную (дорсолатеральную) системы.
Медиальную систему составляют ретикулоспинальные, вестибулоспинальные, тектоспинальный и интерстициоспинальный пути. К медиальной системе относят также нисходящие норадренергические волокна, следующие от голубого пятна, и серотонинергические волокна, следующие от понтомедуллярного ядра шва и подавляющие активность сгибателей. Патология данной системы и формирует постуральную ригидность и «спинальную» форму паркинсонизма. Латеральная система включает в себя прежде всего руброспинальный путь, а также волокна от вентролатеральной части покрышки моста и ядер шва нижних отделов ствола — базисное проявление патологии — рубральный тремор.
Вегетативная нервная система (systema nervosum autonomicum) (ВНС) — это базисная система центральной нервной системы, на которой и разворачивается многоликая экстрапирамидная патология, включая не только болезнь Паркинсона. ВНС разделяется на надсегментарный и сегментарный отделы НС.
Центральный (надсегментарный) отдел ВНС
Предцентральные и лобные области включают в себя моторные центры иннервации неисчерченных (гладких) мышц внутренних органов и сосудов, центры рецепции из внутренних органов и сосудов, центры потоотделения и др. Полосатое тело — центры терморегуляции, слюно- и слезоотделения. Ядра ретикулярной формации — надсегментарные центры функций дыхания, вазотонус, сердечную деятельность, глотание и др. Мозжечок участвует в вегетативной иннервации зрачка.
Периферический отдел ВНС представлен нервами и узлами, расположенными вблизи внутренних органов (экстрамурально) либо в их толще (интрамурально).
Рефлекторная дуга ВНС. I нейрон — афферентный (чувствительный) — расположен в спинномозговых узлах и в узлах черепных нервов. II нейрон — вставочный — расположен в сегментарных вегетативных центрах. III нейрон — эфферентный — расположен в вегетативных узлах.
Нейромедиаторы ВНС
Ацетилхолин (АХ) — медиатор преганглионарных нейронов обоих отделов ВНС, а также медиатор постганглионарных парасимпатических нейронов и симпатических нейронов, иннервирующих потовые железы. Норадреналин (НА) — медиатор симпатических постганглионарных нейронов.
Мозговой слой надпочечников выделяет в кровоток адреналин (А) под влиянием холинергической регуляции симпатической нервной системы.
Классификация заболеваний ВНС, разработанная Американским обществом по изучению автономной нервной системы (2010)
Надсегментарный уровень ВНС:
- катехоламиновые расстройства;
- центральные вегетативные расстройства:
- изолированная (чистая) вегетативная недостаточность (синдром Брэдбери — Эгглстона);
- болезнь Паркинсона;
- ортостатическая гипотензия;
- синдром постуральной тахикардии;
- мультисистемная атрофия (синдром Шая — Дрейджера);
- семейная дизавтономия (синдром Рейли — Дея);
- диабетические вегетативные нарушения.
Для вегетативных нарушений характерна клиническая характеристика двух типов нейродегенеративных расстройств, обусловленных дисфункцией 2 типов белков: тау-белка и альфа-синуклеина, что и приводит к таупатиям и синуклеинопатиям в рамках нозологических форм.
Тау-белок (таупатии)
Внутриклеточные включения, обычно выявляющиеся в нейронах с помощью специального окрашивания и состоящие из скрученных или прямых нитевидных структур (нейрофиламентов). Различные по структуре и химическому составу нейрофибриллярные включения (клубочки), образованные избыточно фосфорилированным тау-белком.
Альфа-синуклеин
Синуклеины — семейство белков, обнаруживаемых в нервной ткани и в некоторых видах опухолей. Известно три формы белка: альфа-синуклеин, бета-синуклеин, гамма-синуклеин. Альфа-синуклеин обнаруживается в нервных окончаниях и составляет около одного процента общего белка мозга. Бета-синуклеин экспрессируется в основном в мозге и локализуется в пресинаптических нервных окончаниях. Гамма-синуклеин выявляется преимущественно в периферической нервной системе.
При БП и других заболеваниях обнаруживаются цитоплазматические включения, называемые тельцами Леви, основной компонент которых представлен фибриллярной формой белка альфа-синуклеина. В то же время тельца Леви обнаруживаются в нервных клетках и при других нейродегенеративных заболеваниях, таких как деменция с образованием телец Леви, множественная системная атрофия, фронтотемпоральная дегенерация, на основании чего БП и перечисленные заболевания объединяют в одну нозологическую группу, получившую название «синуклеопатии».
К нейродегенеративным экстрапирамидным заболеваниям относятся (по Mackenzie et al., 2010):
1. Таупатии:
— прогрессирующий супрануклеарный парез взора;
— кортикобазальная дегенерация;
— болезнь серебряного зерна;
— фронтотемпоральная деменция и паркинсонизм 17-й хромосомы (FTDP-17);
— болезнь Альцгеймера;
— болезнь Пика.
2. Синуклеопатии:
— болезнь Паркинсона;
— деменция с тельцами Леви;
— мультисистемная атрофия;
— TDP-43-протеинопатия;
— дегенерация фронтотемпоральных долей с TDP-43 (FTLD-TDP).
Кроме этого, выделяют нейродегенеративные заболевания с патологией других белков:
1. Фузопатии:
— дегенерация фронтотемпоральных долей с FUS (FTLD-FUS);
— neuronal intermediate filament inclusion disease (NIFID);
— basophilic inclusion body disease (BIBD).
2. Тринуклеотидные заболевания:
— хорея Хантингтона;
— спинобульбарная мышечная атрофия, тип Кеннеди;
— атаксия Фридрейха;
— спиноцеребеллярная атаксия;
— дендаторубро-паллидолуизальная атрофия (DRPLA).
3. Прионные заболевания protein ifection (PRION):
— болезнь Крейтцфельдта — Якоба;
— синдром Герстмана — Штраусслера — Шейнкера;
— фатальная семейная бессонница;
— куру;
— заболевания, которые все чаще встречаются в практике невролога.
4. Нейроаксональные дистрофии:
— инфантильная нейроаксональная дистрофия;
— нейродегенерация с отложением железа в мозге.
5. Заболевания мотонейрона:
— боковой амиотрофический склероз;
— первичный боковой склероз;
— спинальная мышечная атрофия.
Сегодня установлена связь семейного БАС с экспансией интронной последовательности повторов гексануклеотидов в C9orf72. Эти обстоятельства дали основание пересмотреть концепцию течения БАС и фронтотемпоральной деменции на основании общей патогенетической сущности в виде цитоплазматических включений убиквитированного белка TDP 43.
Таким образом, роль определенных генетически детерминированных белков в патогенезе нейродегенеративных заболеваний (каким и является болезнь Паркинсона и его фенокопии) очевидна. Не исключено, что ключ в терапии подобных заболеваний лежит в найденном способе торможения функционирования данных белков.
Клинические проявления центральных вегетативных расстройств не всегда укладываются в определенную нозологическую форму (от «холодных» или «красных» рук до паркинсонизма):
— ортостатическая гипотензия;
— фиксированный пульс (тахикардия в покое и не учащающийся при вставании пульс);
— артериальная гипертензия в положении лежа;
— гипогидроз;
— гастропарез;
— недержание мочи;
— запор;
— ухудшение зрения в сумерках;
— апноэ во сне.
И все же диагностика болезни Паркинсона и заболеваний, ее имитирующих, на сегодняшний день лежит в плоскости знания клиники подобных заболеваний, в частности проявления двух основных экстрапирамидных синдромов.
Основные экстрапирамидные синдромы
1. Гипокинетические синдромы:
1.1. Акинетико-ригидный синдром.
1.2. Изолированная акинезия (без ригидности).
2. Гиперкинетические синдромы:
2.1. Тремор.
2.2. Дистония.
2.3. Хорея.
2.4. Атетоз.
2.5. Баллизм.
2.6. Миоклония.
2.7. Тики.
2.8. Акатазия.
2.9. Пароксизмальные дискинезии.
2.10. Гиперэксплексия.
2.11. Синдром беспокойных ног.
2.12. Стереотипии.
2.13. Синдром ригидного человека.
2.14. Гиперкинезы, индуцированные периферическими (миогенными и невральными) факторами.
2.15. Психогенные гиперкинезы.
Нейрохимические нарушения при первичном паркинсонизме — проявление генетической дегенерации:
— уменьшение синтеза дофамина;
— увеличение уровня ацетилхолина;
— увеличение количества глутамата и аспартата, исходя из дефицита нейромедиаторов, и формирование синдромов экстрапирамидной патологии;
— уменьшение количества норадреналина, серотонина способствует проявлению психических расстройств.
Эти данные характеризуют истинную частоту экстрапирамидной патологии, характерной для Европы и США. В то же время для Украины характерны два вида экстрапирамидной патологии: болезнь Паркинсона и сосудистый паркинсонизм. Учащение фенокопий болезни Паркинсона очевидно, и задачей клиницистов уже при первичных проявлениях в рамках различных вегетативных расстройств является заподозрить другие болезни.
Болезнь Паркинсона — это болезнь «полных суток», и ее симптомы проявляются с момента, когда больной отправляется ко сну, и до момента, когда больной просыпается и встает с постели, и являются неотъемлемой частью клинической картины заболевания. Но уже в начальной стадии болезни Паркинсона необходимо обратить внимание на моторные и немоторные проявления этого дегенеративного заболевания.
1. Моторные проявления:
— нарушение походки (брадикинезия, брадифрения, замедленный старт-рефлекс, топтание на месте);
— изменение речи (монотонность, брадилалия, дисфония);
— нарушение глотания.
Уже на этих этапах проверяется симптом Нойка. Обращают внимание на гипомимию и другие симптомы, ведь симптом «зубчатого колеса» — далеко зашедшие стадии паркинсонизма.
У 20 % больных БП ночью возникают крампи — болезненные непроизвольные мышечные спазмы, чаще в мышцах голеней и стоп, реже — в двуглавой мышце плеча или разгибателей пальцев кисти, продолжающиеся от нескольких секунд до нескольких минут. В большинстве случаев крампи наблюдаются в покое; они могут быть спровоцированы интенсивной физической нагрузкой, приемом алкоголя, недостатком сна, курением, переохлаждением или перегреванием, нарушением водно-электролитного баланса. У 10 % больных БП выявляется ночная миоклония — кратковременные мышечные подергивания, обычно во время медленного сна, на фоне длительного лечения леводопой, чаще у больных с дневными дискинезиями. Основными причинами развития моторных флуктуаций, в том числе ночной акинезии при БП, являются колебания концентрации леводопы в плазме крови из-за короткого периода полужизни двухкомпонентных препаратов (леводопа/ингибитор ДДК), что, в свою очередь, ассоциируется с пульсирующей нефизиологической стимуляцией дофаминовых рецепторов.
2. Немоторные проявления болезни Паркинсона:
— эмоциональные расстройства — снижение памяти, депрессия, панические атаки;
— вегетативные нарушения:
— дисфункция желудочно-кишечного тракта (стойкие запоры);
— нарушения мочеиспускания (учащение и императивность мочеиспускания), никтурия, неполное опорожнение мочевого пузыря — первый симптом БП (остаточная моча в пузыре);
— нарушение половой функции (эректильная дисфункция при наличии гиперсексуальности);
— дисфункция сердечно-сосудистой системы:
— ортостатическая гипотензия (после 3-минутного стояния систолическое давление снижается более чем на 20 мм рт.ст., а диастолическое — более чем на 10 мм рт.ст.) может быть причиной синкопальных состояний, головокружения, головной боли. Измерение давления стоя;
— нарушение потоотделения;
— гипогидроз, вызывающий непереносимость высокой температуры воздуха;
— похудение;
— нарушение слюноотделения;
— сальность кожи;
— себорейный дерматит.
Три этапа диагностики экстрапирамидного расстройства
1. Распознавание экстрапирамидного синдрома.
2. Уточнение анамнестических данных; выявление сопутствующих синдромов; лабораторные исследования; определение нейровизуализационных маркеров (СКТ, МРТ, ПЭТ); леводопа-диагностика; ЭНМГ-ди-агностика.
3. Установление нозологического диагноза (при паркинсонизме определение степени тяжести по рейтинговым шкалам).
Таким образом, применение карты Well-Being Мар (WBM-PD8) для оценки самочувствия больных БП дает возможность считать ее высокоинформативным инструментом для анализа субъективного состояния больных в их повседневной жизни и может применяться в качестве объективного показателя качества жизни пациентов. Применение карты WBM-PD8 в повседневной практике врача-невролога объективно отражает темп прогресса нейродегенеративного процесса при БП и эффективность комплексной патогенетической терапии.
Диагностические критерии
а) наличие гипокинезии (замедленность инициации произвольных движений с прогрессирующим снижением скорости и амплитуды повторных движений);
б) наличие по меньшей мере одного из следующих симптомов:
— мышечная ригидность;
— тремор покоя 4–6 Гц;
— постуральная неустойчивость, не связанная со зрительной, вестибулярной, мозжечковой или проприорецептивной дисфункцией.
Диагностический тест с леводопой
Осуществляется прием суточной терапевтической дозы препарата, содержащего леводопу (наком, L-допа, мадопар), 200–250 мг в течение 4–5 дней. Во избежание внетерапевтического действия леводопы за час до ее приема назначается 10 мг мотилиума.
Не забывать: нарушение обоняния — первый симптом при болезни Альцгеймера, но достаточно часто при синдроме и болезни Паркинсона!
Нарушение глотания — четкий признак экстрапирамидной патологии, который особенно характерен для сосудистого паркинсонизма и кортикобазальной дегенерации (КБД). Рентгенологический тест с барием в дифференциальной диагностике крайне важен. Диагностируется локальная мышечная ригидность: глотка, пищевод, желудок.
Критерии исключения болезни Паркинсона
— Анамнестические указания на повторные инсульты со ступенеобразным прогрессированием симптомов паркинсонизма, повторные черепно-мозговые травмы или достоверный энцефалит.
— Лечение нейролептиками перед дебютом болезни.
— Строго односторонние проявления в течение более 3 лет.
— Супрануклеарный паралич взора.
— Мозжечковые знаки.
— Раннее появление симптомов выраженной вегетативной недостаточности:
— раннее появление выраженной деменции;
— высокие рефлексы.
Базисные критерии, подтверждающие болезнь Паркинсона
— Одностороннее начало проявлений болезни.
— Наличие тремора покоя.
— Постоянная асимметрия с более выраженными симптомами на стороне тела, с которой началась болезнь.
— Хорошая реакция (70–100 %) на Л-ДОФА.
— Прогрессирующее течение заболевания.
— Наличие выраженной дискинезии, индуцированной Л-ДОФА.
— Откликаемость на Л-ДОФА в течение 5 лет и более.
— Длительное течение заболевания (10 лет и более).
Для клинической оценки тяжести болезни Паркинсона используются стандартные международные шкалы: UPDRS, MDS-UPDRS, шкала повседневной жизненной активности Schwab и England, модифицированная шкала Hoehn и Yahr.
Степени тяжести болезни Паркинсона по Хен — Яру (Hoehn — Yahr):
0 — нет симптомов болезни;
1 — унилатеральное проявление;
2 — билатеральное проявление без нарушения равновесия;
3 — от умеренной до выраженной степени тяжести; некоторая постуральная неустойчивость, но не нуждается в посторонней помощи; необходима поддержка для удержания равновесия при выполнении теста на постуральную устойчивость;
4 — тяжелая инвалидность, но может самостоятельно ходить и стоять;
5 — прикован к креслу или кровати, если нет посторонней помощи.
Принципы назначения терапии больным паркинсонизмом
— При установлении диагноза БП больной должен непрерывно принимать противопаркинсонические средства (ППС).
— Лечение начинается в виде монотерапии одним из ППС.
— Препаратом выбора может быть любое ППС (при отсутствии противопоказаний), в том числе холинолитик.
— Монотерапия начинается с субпороговых доз, оптимальная доза подбирается постепенно, в течение не менее 3–4 недель, и осуществляется в пределах фармакотерапевтического окна с установлением оптимальной однократной дозы и кратности приема.
— Комбинированная терапия назначается при недостаточной эффективности монотерапии с добавлением одного ППС в субпороговой дозе с последующим постепенным увеличением дозы.
— Длительное лечение БП препаратами леводопы со временем приводит к возникновению клинического патоморфоза, требующего специальной коррекции.
Основные лекарственные средства в лечении болезни Паркинсона и его фенокопий
Проблема лечения болезни Паркинсона, а тем более его фенокопий, крайне затруднительна.
Вот почему (после установления достоверного диагноза) следует определиться прежде всего со степенью тяжести клинических проявлений, но особенно обратить внимание на противопоказания при назначении лекарственных средств. У каждого больного они свои!
В настоящее время одним из наиболее значимых и эффективных представителей АДР, имеющим обширную и весомую доказательную базу, является прамипексол — орион (мирапекс), применяемый с 1996 г. для терапии ранних и поздних стадий БП. Прамипексол представляет собой неэрголиновое производное аминобензотиазола. Назначение: 1-я неделя — 0,125 мг 3 р/сут, 2-я неделя — 0,125 мг 4 р/сут, далее 0,125 мг 5–6 р/сут — общая суточная 1,5 мг.
У пациентов с далеко зашедшей стадией БП прамипексол (в комбинации с препаратами левоподы) также активен в отношении основных двигательных проявлений паркинсонизма, но одновременно дает возможность:
— сократить общую длительность периодов «выключения» (в среднем на 30–40 %);
— увеличить период «включения» (в среднем на 2 ч в сутки);
— снизить суточную дозу леводопы (на 15–30 %) и таким образом уменьшить тяжесть леводопаиндуцированных дискинезий.
Отдельно следует отметить влияние прамипексола на тремор — симптом, достаточно резистентный к традиционной противопаркинсонической терапии.
Еще одно специальное показание к назначению прамипексола — аутосомно-рецессивный ювенильный паркинсонизм.
Потенцирование эффекта леводопасодержащих препаратов
Назначают дермопласт неупро, который содержит препарат ротиготин, существенно потенцирующий эффект леводопы (2–4–8 мг). Действует в течение суток, улучшает как моторные, так и немоторные симптомы болезни Паркинсона, возможно применять при начальных проявлениях БП в монотерапии, медленно, повышая дозу до 16 мг, а также применять как диагностический тест. Не сочетать с антидепрессантами, алкоголем, не применять при тяжелой патологии печени.
Ротиготин рассматривают как специфический агонист дофаминовых рецепторов с преобладанием агонизма к D3-рецепторам над агонизмом к D2- и D1-рецепторам. По сравнению с дофамином ротиготин в 2600 раз сильнее в отношении D3-рецепторов и примерно в 20 раз сильнее остальных подтипов ДА-рецепторов.
Главное преимущество трансдермальной формы ротиготина (неупро) состоит в транспорте препарата вне его взаимодействия с пищей и другими лекарственными средствами для перорального приема. Пластырь наклеивается на кожу один раз в сутки, что значительно облегчает соблюдение режима терапии.
В течение последних нескольких десятилетий для лечения больных БП применяются двухкомпонентные препараты, содержащие леводопу и ингибитор периферической ДОФА-декарбоксилазы (ДДК), вызывающий торможение периферического декарбо-ксилирования леводопы. С 2003 года для лечения БП применяется трехкомпонентный препарат — сталево, содержащий леводопу, карбидопу и ингибитор катехол-О-метилтрансферазы энтакапон.
Вместе с тем леводопа имеет и другой путь проникновения в организм (Евтушенко С.К., Неймарк Е.З. Трансцеребральный назальный электрофорез с леводопой в терапии болезни Паркинсона. Авторское свидетельство, 1980 г., Москва).
И все же самым перспективным методом лечения болезни Паркинсона на сегодняшний день является применение подкожной автоматически управляемой дофаминовой помпы (Карабань И.Н., 2012).
Побочные явления терапии болезни Паркинсона
1. Ранние (первые дни и недели лечения дофаминомиметиками):
— снижение аппетита;
— тошнота, рвота;
— ортостатическая гипотензия.
2. Поздние (3–9 лет от начала терапии):
— снижение порога чувствительности к некоторым побочным эффектам препарата;
— снижение эффективности терапии;
— ухудшение когнитивных функций;
— возникновение психических нарушений.
— синдром on-off.
Лекарственные дискинезии:
1. Дискинезии на пике дозы (on-дискинезии):
— хореические;
— дистонические;
— баллистические насильственные движения.
2. Двухфазные дискинезии (в начале и конце действия препаратов леводопы):
— хореические;
— дистонические;
— баллистические насильственные движения;
— смешанные.
3. Дистония of-периода (дистония раннего утра):
— спастические дистонические позы;
— преимущественно в ногах;
— провоцируются движением.
4. Акатизия (неспособность длительно оставаться в покое из-за непреодолимого желания находиться в постоянном движении).
5. Стереотипии.
Вместе с тем, кроме базисной, больные нуждаются и в симптоматической терапии. В частности, на высоте достижения эффекта от леводопасодержащих препаратов возможно применение гамалате В6 в целях предотвращения синдрома on-off и воздействия на ларвированную депрессию, головные боли. Целесообразно назначение гамалате В6 по 1 табл. 3 р/день, длительностью до 1 месяца, с перерывами, с учетом жалоб больного. При снижении памяти — препараты гинкго билобы.
При терапии леводопа-содержащими препаратами больше 6 месяцев — применение препаратов с антиоксидантной и мембранотропной функцией, которые уменьшают глутаматную эксайтотоксичность, восстанавливают нейромедиаторный баланс. Применение мексидола целесообразно, тем более мексидол оказывает действие на нарушенные когнитивные функции как при БП, так и при естественном старении.
Следует быть осторожными при постановке диагноза БП, если у больного:
— симметричность симптоматики — отсутствие тремора;
— раннее появление постуральной неустойчивости;
— преобладает нарушение ходьбы над брадикинезией в руках,
— преобладание аксиальной ригидности над ригидностью конечностей (высокий тонус мышц шеи по сравнению с тонусом в руках);
— отсутствие ответа на препараты леводопы.
Начало паркинсонического синдрома характерно для таких нейродегенераций:
— Деменция с тельцами Леви.
— Кортикобазальная дегенерация.
— Прогрессирующий супрануклеарный паралич.
— Мультисистемная атрофия.
Подкорковые нейродегенерации:
— ДТЛ, развивающаяся в течение года до начала моторных нарушений, но в дебюте.
— Зрительные цветные галлюцинации еще до начала применения дофаминергической терапии. Галлюцинации исчезают после представления объекта, который ему кажется.
— Медленное, в течение 2–3 месяцев, нарастание скованности движений.
— Вегетативные нарушения (гипогидроз, изолированная акинезия).
— Присоединение брадикардии.
— Экстрапирамидный синдром без асимметрии.
Кортикобазальная дегенерация:
Дебют:
— апраксия,
— афазия,
— корковые нарушения чувствительности (лицо — рука),
— непроизвольные движения или «левитация конечностей» — феномен «чужой руки» («она не моя»), брадикинезия, брадифрения,
— на МРТ — атрофия теменных долей мозга,
— синдром «чужой руки» — многие неврологи этот феномен не представляют. Если вы смотрели последнюю часть киноэпопеи о Гарри Поттере, то можете вспомнить, как одного из героев по имени Хвост задушила его же собственная рука. Синдром «чужой руки» и есть облигатное проявление КБД.
Особенно важным в диагностике КБД является подтверждение локальных нарушений, в частности, мочевыделительной системы, при этом применяется специальный метод ЭМГ наружного сфинктера мочеточника или анального сфинктера, который выявляет денервационные изменения в них из-за поражения сакрального ядра Онуфа, откуда иннервируются эти мышцы.
Прогрессирующий супрануклеарный паралич Ричардсона — Ольшевского (ограничение движений глазных яблок по горизонтали, позже — по вертикали). Частая жалоба — затруднение взгляда вниз, «застывший взгляд»:
— прогибание шеи и туловища в отличие от «сгибательной» позы при БП — «гордец», симптом «грязного галстука»,
— дизартрия,
— дисфагия (псевдобульбарный синдром),
— позднее развитие деменции,
— на МРТ — атрофия среднего мозга.
Мультисистемная атрофия (МСА) — комбинация экстрапирамидных, пирамидных, мозжечковых и вегетативных расстройств, включая нистагм, адиадохокинез, положительный симптом Нойка:
— постуральная гипотензия,
— падения,
— нарушение мочеиспускания,
— тревога,
— запоры,
— нарушение потоотделения,
— дизартрия,
— дисфагия,
— деменция,
— на МРТ — атрофия мозжечка и моста.
Выделяют три типа МСА:
1. Оливопонтоцеребеллярный — мозжечковые атаки.
2. Стрионегральные признаки паркинсонизма.
3. Синдром Шая — Дрейджера — признаки прогрессирующей вегетативной недостаточности: брадикинезия, гипотония и падения (обмороки). Дифференцируют с другим симптомом Шая — Дрейджера, обусловленным слабостью синусового узла: тяжелые обмороки и падения.
Мультисистемная атрофия
В 1960 г. два исследователя — Milton Shy и Glen Drager описали комплекс неврологических нарушений, ассоциирующихся с вегетативными расстройствами, в настоящее время известный как мультисистемная атрофия. Это спорадическое прогрессирующее заболевание с поздним началом характеризуется вегетативной дисфункцией, синдромом паркинсонизма и атаксией в различных комбинациях.
Клиническая картина
Обычно мультисистемная атрофия развивается между 5-й и 6-й декадами жизни, мужчины страдают несколько чаще женщин (соотношение составляет 1,3 : 1).
Самый частый моторный синдром — синдром паркинсонизма. Он обладает рядом клинических особенностей, позволяющих уже в дебюте заболевания проводить эффективную дифференциальную диагностику с болезнью Паркинсона. Синдром паркинсонизма часто начинается асимметрично, как и болезнь Паркинсона.
Клинически заподозрить мультисистемную атрофию позволяют так называемые симптомы «красного флага»:
— ранние глазодвигательные нарушения;
— низкий ответ на леводопу;
— фокальная дистония (антеколлис);
— ранние постуральные нарушения;
— быстрое клиническое прогрессирование симптомов;
— фокальный миоклонус;
— феномен Рейно, или акроцианоз;
— дисфагия;
— усиление храпа, сонные апноэ;
— насильственный (псевдобульбарный) плач или смех;
— контрактуры.
Кроме того, для МСА характерны:
— нарушение дыхания — связаны с гипокинезом мышц грудной клетки и диафрагмы, дисфункцией мышц верхних дыхательных путей;
— нарушения обоняния, вкуса;
— соматосенсорные расстройства (боль, ощущения покалывания, жжения, зуда, онемения).
Психические нарушения и когнитивные расстройства при МСА:
— замедленность мыслительных процессов (брадифрения);
— нарушение внимания;
— ограничение способности к запоминанию и активному воспроизведению вербальной и зрительной информации;
— зрительно-пространственные нарушения (ограничение способности копировать и воспроизводить по памяти рисунки и фигуры);
— больные производят впечатление аутичного человека на фоне экстрапирамидной симптоматики.
Вызванные потенциалы головного мозга: метод выделения изменений электрической активности мозга в ответ на предъявляемый стимул.
Метод вызванных потенциалов:
— объективный,
— неинвазивный,
— пациент не может произвольно изменить результат,
— быстрота исследования.
Использование различных паттернов и модальностей позволяет исследовать функции практически всех областей мозга.
Модальность вызванных потенциалов: слуховые и зрительные.
Зрительные вызванные потенциалы на реверсию шахматного паттерна:
— реверсивный черно-белый высококонтрастный паттерн — парвоцеллюлярная система нейронов (тип I) затылочной и височной коры;
— низкоконтрастный паттерн — магноцеллюлярная система нейронов затылочной и теменной коры;
— равнояркий цветовой (красно-синий и красно-зеленый) паттерн — парвоцеллюлярная система нейронов (тип II) затылочной коры и зоны V4;
— квадранты зрения.
Когнитивные расстройства можно классифицировать на основе их связи с нарушением в стратегических когнитивных зонах и диагностировать согласно отклонениям от нормы соответствующих этим зонам параметров ВПСС, но вместе с тем клиника в диагностике МСА является ведущей.
Нарушения бодрствования при МСА:
— инсомния;
— парасомнии (психомоторное возбуждение во сне, ночные кошмары, сноговорение, панические атаки) — может появляться до основных моторных симптомов. Гиперсомния (дневная сонливость) — на фоне «легкого» паркинсонизма.
И все же диагноз МСА возможно не только заподозрить, но и поставить, обратив внимание на базисные симптомы ее проявления (Евтушенко С.К., Симанов Р.В. Мультисистемная атрофия: клиника, диагностика, лечение // МНЖ. — 2012. — № 5(51). — С. 42-48).
Лечение МСА
— Дермопласт — неупро exuvantibus.
— Дельталицин (семакс) — капли в нос по схеме 10 дней 1 раз в три месяца.
— Препараты гинкго билобы по 1 капс. 3 р/день до 3 месяцев + когнум.
Но при инсомнии (или парасомнии) — формула сна (фитопрепарат + МgВ6) по 1 табл. + мелатонина 1 табл. на ночь.
При тревоге — гамалате В6.
Для предотвращения падений рекомендовать трость, наколенники.
Сосудистый паркинсонизм
I. Болезнь малых сосудов головного мозга:
— гипертоническая микроангиопатия (липогиалиноз);
— сенильная микроангиопатия (сенильный артериолосклероз, сенильная извитость артерий);
— васкулиты и васкулопатии (узелковый полиартериит, ангиит ЦНС, СКВ);
— наследственные артериопатии.
II. Поражение крупных мозговых артерий:
— атеросклероз крупных (экстра- и интракраниальных) артерий;
— менинговаскулярный сифилис.
III. Кардиогенные поражения головного мозга:
— кардиогенные эмболии;
— гипоксическая энцефалопатия (некроз базальных ганглиев).
IV. Другие заболевания:
— артериовенозные мальформации;
— антифосфолипидный синдром;
— коагулопатии.
Сложившимся частым заблуждением неврологов является то, что сосудистый паркинсонизм встречается до 40 %. Мировая статистика говорит, что — только до 13 %. Следовательно, длительное лечение СП продолжает быть неэффективным.
В чем причина?
Аксиома: церебральная сосудистая дисфункция проявляет генетическую «предуготованность» (Евтушенко С.К. Леводопа и мидантан в лечении болезни Паркинсона // Журнал неврологии и психиатрии им. С.С. Корсакова. — 1974).
Генетические факторы развития болезни Паркинсона
— PARKI (α-синуклеин). Белок альфа-синуклеин играет важную роль в синаптическом везикулярном транспорте и хранении нейротрансмиттеров. Мутации (наследственные или вследствие воздействия экзогенных нейротоксических факторов) в гене α-синуклеина приводят к изменению структуры белка, его накоплению в нейроне и агрегации с образованием телец Леви. В настоящее время α-синуклеин рассматривается в качестве ключевого молекулярного маркера патологии нейронов и модуляции процессов нейродегенерации «паркинсонического» типа.
— PARK2 (паркин). Мутации в гене паркина являются частой причиной раннего, в том числе ювенильного, паркинсонизма (до 50 % семейных форм и около 15 % спорадических случаев). Паркин представляет собой убиквитинпротеинлигазу типа Е3, функция которой заключается в доставке аномально конформированных белков в протеасомный комплекс для последующего расщепления.
— PARK6 (PINKI). Форма аутосомно-рецессивного паркинсонизма обеспечивает развитие до 9 % случаев раннего начала заболевания. Белок PINKI является митохондриальной протеинкиназой и играет важную роль в митохондриальном биогенезе.
— PARK7 (DJ-I). Редкая форма аутосомно-рецессивного паркинсонизма — 1–2 % ранних случаев БП. Белок DJ-I играет важную роль в поддержании целостности и выживаемости дофаминергических нейронов.
— PARK8 (LRRK2). Ген связан с аутосомно-доминантной формой паркинсонизма с пенетрантностью до 40 %. Ген LRRK2 имеет большое значение в развитии спорадических случаев первичного паркинсонизма: от типичной поздней БП с тельцами Леви до атипичных вариантов синуклеин- и тау-патологии. Белковым продуктом гена является дардарин, именно его патологическая активация является следствием доминантной мутации в гене LRRK2 и приводит к развитию нейродегенеративных изменений.
— Ген GBA. Кодирует лизосомальный фермент глюкоцереброзидазу. Мутации в указанном гене могут сопровождаться развитием различных вариантов синуклеинопатий — классической БП и деменции с тельцами Леви.
Клинические особенности паркинсонического синдрома при сосудистом паркинсонизме:
— двустороннее начало заболевания и относительная симметричность симптоматики;
— отсутствие тремора покоя;
— низкая эффективность дофаминергических средств;
— преобладание симптоматики в аксиальных отделах и нижних конечностях;
— раннее развитие постуральных нарушений и изменений ходьбы;
— отсутствие флуктуаций и дискинезий при длительном приеме леводопы.
Сопутствующие синдромы СП:
— раннее развитие псевдобульбарного синдрома;
— раннее развитие нейрогенных нарушений мочеиспускания;
— раннее развитие деменции;
— пирамидный синдром;
— лобные знаки (хватательный рефлекс, паратонии);
— мозжечковая атаксия;
— другие экстрапирамидные синдромы (гемидистония, миоклония);
— очаговые нарушения высших мозговых функций (афазия, апраксия и др.).
Типичный вариант СП:
— акинетико-ригидный синдром (симметричный или асимметричный);
— большая выраженность симптомов в нижних конечностях (шаркающая походка);
— дебют с нарушения ходьбы (но не падения);
— отсутствие эффекта от леводопы.
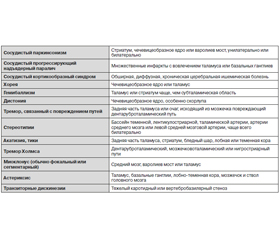
Нейровизуализационные изменения при СП:
— множественные лакунарные инфаркты в базальных ганглиях, стволе, глубинных отделах белого вещества;
— диффузные поражения белого вещества:
— субкортикальный сливающийся или частично сливающийся лейкоареоз;
— распространенный перивентрикулярный лейкоареоз с неровными контурами, распространяющийся в субкортикальную область;
— территориальные подкорковые инфаркты в базальных ганглиях и прилегающем белом веществе;
— двусторонние (реже односторонние) территориальные инфаркты лобных долей;
— одно-, двусторонние инфаркты в области таламуса;
— геморрагические очаги в базальных ганглиях, среднем мозге, таламусе;
— церебральная атрофия с расширением желудочковой системы и корковых борозд.
Лечение сосудистого паркинсонизма
1. Наком (юмекс) + амантадин длительно с учетом внетерапевтических действий.
2. Холестеринолитики (липримар 20 мг в сутки) длительно.
3. Антигипертензивные средства.
4. Фезам 1 табл. 2 р/день до 3 месяцев.
5. Цикл ангиопротекторов (цитофлавин, кавинтон).
6. Антигипоксиданты (мексидол) курсами.
7. Кардиомагнил 75 мг длительно.
Для лечения деменции в настоящий момент доказана эффективность 4 препаратов с двумя механизмами действия: иХЭ — донепезила, галантамина, ривастигмина, нейромидина и блокатора глутаматных NMDA-рецепторов мемантина. Данные препараты повышают концентрацию ацетилхолина благодаря обратимому ингибированию АХЭ и тормозят — бета-амиолиоида структур мозга.
В завершение лекции следует акцентировать внимание врачей-неврологов: диагноз болезни Паркинсона — прежде всего клинический, а не параклинический!
И все же в этой труднокурабельной неврологической проблеме есть обнадеживающие факты. У достаточного количества пациентов именно с болезнью Паркинсона длительно сохраняются память и когнитивные функции. Этому пример — известные писатели, поэты, политики, церковные служащие. Обнадеживает тот факт, что достоверная диагностика и патогенетическая терапия болезни Паркинсона, а не ее фенокопий и приносят эффект.
Когда следует заподозрить атипичный паркинсонизм?
Моторные симптомы: раннее появление постуральной неустойчивости и падений; быстрое прогрессирование паркинсонизма; отсутствие или нестойкий ответ на терапию леводопой; пирамидные и/или мозжечковые симптомы, раннее появление дизартрии и/или дисфагии.
Основными структурными изменениями при МРТ у больных БП являются: конвекситальная атрофия корковых отделов долей мозга, расширение боковых желудочков. При сосудистом паркинсонизме обнаруживаются: лейкоареоз, лакунарные инфаркты, расширение периваскулярных пространств.
Определение риска возникновения церебральных гиперинтенсивных сосудистых изменений при болезни Паркинсона зависит от наличия в анамнезе артериальной гипертензии, ишемической болезни сердца, сахарного диабета, а также ортостатической гипотензии и гипергомоцистеинемии (при уровне гомоцистеина > 13,5 мкмоль/л).
Более специфичны для МСА появление гиперинтенсивной полоски по наружному краю скорлупы на фоне ее атрофии и снижение сигнала на Т2-ВИ. При оливопонтоцеребеллярной атрофии (как варианте МСА) чаще встречаются инфратенториальные изменения — атрофия моста и мозжечка, изменения интенсивности сигнала от основания моста с формированием так называемого симптома «креста» или «пасхальной булочки».
По мере прогрессирования КБД развивается асимметричная атрофия в лобно-теменной области полушарий и стриатуме — на сторону, контралатеральную пораженным конечностям.
МРТ-картина при сосудистом паркинсонизме не ограничивается очаговыми и/или диффузными изменениями вещества головного мозга. На томограммах всегда выявляется наличие умеренных и выраженных атрофических изменений на фоне прогрессирующего сосудистого поражения мозга. Атрофия полушарий более выражена конвекситально и уменьшается в передне-заднем направлении (от лобных долей к затылочным).
Наиболее частыми вариантами сосудистого паркинсонизма являются стриатопаллидарный и фронтостриатный. При стриатопаллидарном варианте при МРТ наблюдаются, как правило, двусторонние множественные лакуны в базальных ганглиях и/или множественное диффузное расширение периваскулярных пространств проекции лентикулярных ядер.
Для фронтостриатного варианта СП характерными МРТ-изменениями является обширный субкортикальный и перивентрикулярный лейкоареоз в глубинных отделах лобных долей в сочетании с лакунарными инфарктами в базальных ядрах. В случае нигростриатного варианта необходимо наличие очагового поражения среднего мозга, захватывающего черную субстанцию.
Как правило, сосудистый паркинсонизм сочетается с другими признаками органического поражения мозга пирамидными, мозжечковыми, псевдобульбарными, постуральными и когнитивными нарушениями (положительный симптом Нойка уже на начальных стадиях БП).
Наиболее информативным в диагностике БП является протонная магнитно-резонансная спектроскопия. Количественные оценки процентного содержания в ткани отдельных молекул дают важную биологическую информацию, которая может быть использована в клинике при исследованиях роли отдельных метаболитов в условиях нормы и патологии.
N-ацетиласпартат (NAA). МР-сигнал N-ацетил-аспартата, как правило, наиболее интенсивен в спектре ПЭТ.
Холин (Cho). Этот сигнал представляет общее количество запаса холина в мозге, включая ацетилхолин нейромедиатора, фосфохолин и мембранный фосфатидилхолин. Холин образуется главным образом в печени из фосфатидилхолина, синтезируемого из фосфатидил–этаноламина посредством серии реакций метилирования.
Креатин/фосфокреатин (Cr/PCr). Интенсивность обновления энергией фосфорных соединений в головном мозге очень велика. Поэтому содержание АТФ и креатинфосфата в мозговой ткани характеризуется значительным постоянством. Распад аденозинтрифосфата (АДФ) и неорганического фосфата — это основной источник энергии.
Глутамат (Glu). Возбуждающий нейромедиатор. Глутаминовая система занимает ключевую позицию в обменных процессах мозга. Возбуждение нервной системы сопровождается повышением содержания аммиака в нервной ткани. Глутаминовая кислота способна связывать аммиак с образованием безвредного для нервной ткани глутамина. Глутаминовая кислота увеличивает также синтез ацетилхолина и АТФ, способствует удержанию ионов калия во внутриклеточном пространстве и содействует поляризации.
Глутамин (GIn). Продукт реакции глутамата с аммиаком. Глутаминовая кислота в нервной ткани может декарбоксилироваться с образованием γ-аминомасляной кислоты. Γамма-аминомасляная кислота является важным нейромедиатором, выполняющим тормозные функции. Больше всего γ-аминомасляной кислоты обнаружено в сером веществе коры головного мозга, в то время как белое вещество мозга и периферическая нервная система ее почти не содержат.
При первичном паркинсонизме снижение уровня N-ацетиласпартата и повышение концентрации холина, которые приводят к достоверному снижению соотношения NAA/Cho, выявляются в первую очередь в проекции компактной части черной субстанции.
По мере прогрессирования заболевания и расширения границ нейродегенеративного процесса локализация нарушений, выявляемых у больных с БП, расширяется.
При паркинсонизме в рамках мультисистемных нейродегенеративных заболеваний нейровизуализационная картина характеризуется резким снижением метаболизма глюкозы в лентикулярных ядрах!
У больных КБД наблюдается снижение метаболизма глюкозы в лобно-теменных отделах коры и зрительных буграх, также более существенное — в структурах, противоположных пораженной стороне. Для прогрессирующего надъядерного паралича характерно сочетание гипометаболизма в глубоких структурах мозга с гипометаболизмом в лобной и теменной коре.
Наиболее важным ПЭТ-признаком для дифференциальной диагностики БП и паркинсонизма в рамках нейродегенеративных заболеваний является изменение метаболизма глюкозы в лентикулярных ядрах (в первую очередь в скорлупе), которое позволяет судить о состоянии постсинаптического звена дофаминергической системы.
Автор приносит глубочайшую благодарность ведущему специалисту европейского уровня по проблеме экстрапирамидной патологии д.м.н., проф. Ирине Николаевне Карабань за научные и практические советы в современном освещении данной проблемы.

/61.jpg)
/64.jpg)
/64_2.jpg)
/66.jpg)
/70.jpg)
/63.jpg)