Статтю опубліковано на с. 88-96
До клініки дитячої психоневрології ДУ «Інститут педіатрії, акушерства і гінекології НАМН України» часто звертаються батьки пацієнтів, які мають затримку психомовного та соціального розвитку, різноманітні порушення поведінки. Нерідко у дітей з подібними скаргами діагностуються розлади аутистичного спектра (РАС), у тому числі вторинні стосовно генетичних захворювань. За нашими спостереженнями, у таких пацієнтів частим симптомом є збільшення розмірів голови — симптом, що може становити значну цінність для диференціальної діагностики. Відомо, що макроцефалія є достатньо поширеним і гетерогенним патологічним станом, що з’являється при значній кількості різноманітних захворювань. На сьогодні чіткі алгоритми ведення пацієнтів із макроцефалією відсутні як в Україні, так і за кордоном. У нашій роботі ми спробували проаналізувати макроцефалію як прояв порушення розвитку мозку у дітей з розладами аутистичного спектра, а також підходи до диференціальної діагностики даного стану.
Макроцефалія визначається як збільшення потилично-фронтальної окружності голови (ОГ) більш ніж на 2 стандартних відхилення (SD) для даного віку та статі (тобто ≥ 97-го перцентиля). Деякі автори пропонують вважати макроцефалією збільшення окружності голови більше ніж на 2,5 SD (більше 99,6-го перцентиля) [1, 2].
Причиною аномального збільшення розмірів голови може бути збільшення кісток черепа або структур, що знаходяться в черепній коробці (паренхіми головного мозку, крові, ліквору).
Уперше класифікація макроцефалій у дітей була розроблена в 1986 р. DeMyer [3] та доповнена у подальшому Bodensteiner et Chung в 1993 [4] та Williams у 2008 [1]. Класифікація базується на етіології розладу, а також наявності супутніх вад розвитку, порушень метаболізму та змін головного мозку при нейровізуалізації.
Залежно від етіології умовно виділяються такі типи макроцефалії:
1) мегаленцефалія — збільшення паренхіми мозку;
2) гідроцефалія — збільшення вмісту ліквору;
3) гіперостоз — збільшення розмірів кісток черепа [1].
У закордонній літературі терміни «макроцефалія» та «мегаленцефалія» часто використовують як синоніми. Таким чином, під макроцефалією переважно розуміють збільшення розмірів голови за рахунок паренхіми головного мозку [1]. У нашій роботі ми розглянемо переважно макроцефалію як результат збільшення паренхіми головного мозку. Методи нейровізуалізації можуть дати додаткову інформацію для оцінки розмірів головного мозку, однак найбільш зручним і надійним методом діагностики макроцефалії є вимірювання окружності голови.
Вимірювання потилично-фронтальної окружності голови проводиться шляхом накладення сантиметрової стрічки позаду по найбільш виступаючій частині потиличної кістки, а спереду — по лінії надбрівних дуг. Сантиметрова стрічка, що використовується для виміру окружності голови, не повинна розтягуватись. Планове вимірювання окружності голови проводять від народження до трирічного віку [5]. При оцінці окружності голови повинні бути враховані статура, а також етнічна приналежність пацієнта [6].
Вимірювання потилично-фронтальної окружності голови у новонароджених може бути неінформативним до 3–4-ї доби життя за наявності кефалогематом, швів або післяпологових змін конфігурації голови. У дітей більш старшого віку похибки вимірювання можуть виникати внаслідок густого волосся, дефектів або гіпертрофії кісток черепа [7].
Запідозрити в дитини макроцефалію можна за наявності хоча б одного відхилення розміру ОГ і подальшого прогресивного наростання, тобто переходу з одного перцентильного рівня на інший порівняно з віковою нормою, або за наявності приросту ОГ > 2 см/міс для дітей у віці до 6 місяців [8]. При підозрі на макроцефалію необхідний детальний збір анамнезу та проведення фізикального обстеження, при необхідності — проведення нейровізуалізації.
У більшості випадків ретельний збір анамнезу та дані огляду дозволяють встановити причини збільшення розмірів голови у дитини. Наприклад, низький вік гестації при народженні може вказувати на можливий розвиток гідроцефалії. У певних випадках огляд може дати необхідну інформацію для подальшої диференціальної діагностики. Зокрема, наявність на шкірі дитини пігментованих плям кольору кави з молоком потребує виключення нейрофіброматозу 1-го типу та туберозного склерозу.
При обстеженні дитини з макроцефалією слід звернути увагу на такі ключові моменти.
1. Чи насправді розміри голови є збільшеними? Вимірювання потилично-фронтальної окружності голови повинно проводитися ретельно, з урахуванням зазначених вище вимог. Паралельно проводиться вимірювання маси тіла та зросту. Окружність голови може іноді перебувати у межах нормальних перцентилів, але бути аномальною порівняно з масою та зростом.
2. Чи доступні дані кількох послідовних вимірювань окружності голови (у тому числі при народженні)? Чи відбувається послідовне збільшення перцентилів, у межах яких знаходиться окружність голови? Подібне збільшення необов’язково є ознакою патологічного стану та може бути присутнім при доброякісній сімейній гідроцефалії, однак у поєднанні з іншими симптомами може бути ознакою збільшення внутрішньочерепного тиску.
3. Чи присутні у дитини фактори ризику, що можуть бути пов’язані з розвитком макроцефалії, зокрема недоношеність, субдуральні або інтравентрикулярні крововиливи, перенесені менінгіти, вроджені аномалії розвитку, наявність у сімейному анамнезі генетичних синдромів, аутизму, макроцефалії?
4. Чи є у дитини симптоми підвищеного внутрішньочерепного тиску? У випадку наявності симптомів підвищення внутрішньочерепного тиску необхідно провести обстеження за допомогою методів нейровізуалізації, надаючи перевагу магнітно-резонансній томографії (МРТ).
5. Чи є у дитини затримка розвитку, чи були в анамнезі судоми, втрата навичок, зміни поведінки, інші неврологічні проблеми?
6. Виміряйте окружність голови батьків і членів родини. Відсутність у дитини неврологічної симптоматики за наявності збільшеної окружності голови може свідчити про наявність найбільш поширеної причини макроцефалії у дитячому віці — доброякісної сімейної макроцефалії.
7. Під час огляду звертаємо увагу на специфічні ознаки, що можуть вказувати на етіологію макроцефалії:
— дисморфізм;
— порушення росту (наприклад, прискорений ріст тіла при синдромі Сотоса);
— прояви нейрошкірних захворювань (наприклад, нейрофіброматозу або туберозного склерозу);
— грубі риси обличчя або інші прояви хвороб накопичення (наприклад, мукополісахаридозу);
— наявність вогнищевої неврологічної симптоматики, патології очного дна;
— ознаки підвищеного внутрішньочерепного тиску;
— наявність гепатоспленомегалії, що може вказувати на хвороби накопичення або патології системи крові (наприклад, таласемії) [2].
Пренатально макроцефалія діагностується за допомогою ультразвукового дослідження (УЗД) і визначається як збільшення розміру ОГ більше 2 стандартних відхилень або вище 98-го перцентиля для даного гестаційного віку (оцінка гестаційного віку проводиться шляхом вираховування дня від останньої менструації або за довжиною стегнової кістки). Пренатальна діагностика макроцефалії є ускладненою внаслідок невідповідності між пренатальними та постнатальними кривими росту ОГ відносно віку. Вимірювання ОГ плода є орієнтовними, оскільки у літературі відсутні чіткі нормативні показники з урахуванням статі, раси або етнічної приналежності.
Підхід до оцінки внутрішньоутробної макроцефалії включає врахування інших фетальних біометричних параметрів (наприклад, довжина кісток, окружність тулуба) відносно гестаційного віку, анамнестичних даних (сімейна макроцефалія, споріднені шлюби) та вимірювання ОГ у батьків, братів і сестер. За наявності інших ультрасонографічних аномалій плода (наприклад, дисгенезія мозолистого тіла, вади розвитку кори, гіпертелоризм, збільшення нирок, полідактилія, недорозвиток кісток) можна думати про синдромальну макроцефалію. Більша, ніж необхідно для даного гестаційного віку, ОГ, окружність живота і довжина трубчастих кісток можуть вказувати на синдроми передчасного розвитку (Сотоса, Уївера). Окружність голови у межах 2–2,5 SD для даного гестаційного віку, але без наявних стигм дизембріогенезу може свідчити про сімейну макроцефалію. При підозрі на внутрішньоутробну макроцефалію доцільно провести каріотипування та МРТ головного мозку плода [8].
У випадку пренатально діагностованої макроцефалії проводиться вибір тактики ведення вагітності та пологів. Кесарський розтин показаний у випадках ОГ плода ≥ 40 см. За наявності перинатальної макроцефалії фізіологічні пологи можуть становити небезпеку внаслідок невідповідності між розмірами таза вагітної та розміром голови плода [9].
При постнатальній діагностиці макроцефалії необхідно перш за все диференціювати доброякісні стани, що не потребують додаткового лікування, від макроцефалії, що є проявом певних захворювань. У більшості випадків при скаргах батьків на збільшення розмірів голови у дитини діагностуються доброякісна, або так звана сімейна макроцефалія [1].
Несиндромальна, або доброякісна, макроцефалія є станом, при якому збільшення розмірів головного мозку не пов’язане з будь-якими іншими вадами розвитку або серйозними патологічними станами. Можуть бути наявними певні зміни рис обличчя, вторинні відносно збільшення розмірів голови. Ці зміни можуть включати виступаючий або високий лоб, доліхоцефальну форму голови, іноді — помірний гіпертелоризм, трикутну форму обличчя [1].
Макроцефалія у складі специфічних генетичних синдромів зазвичай супроводжується затримкою фізичного або психічного розвитку, порушеннями поведінки або наявністю супутніх вад розвитку. Сукупність специфічних вад розвитку формує специфічний фенотип, що дозволяє запідозрити певний генетичний синдром. Однак слід відрізняти генетичні синдроми, що обов’язково супроводжуються макроцефалією, від таких синдромів, при яких макроцефалія присутня іноді або рідко [1, 2].
Доброякісна сімейна макроцефалія — найбільш поширена причина макроцефалії у дітей [10]. Діти, у яких діагностується даний стан, народжуються з окружністю голови у межах високих нормальних перцентилів та нормальним розміром тулуба і кінцівок. Протягом першого року життя ОГ збільшується до розмірів, більших за 2 стандартних відхилення. ОГ може збільшуватися від 0,6 до 1 см на тиждень (порівняно з нормальним 0,4 см/тиждень) [11]. Швидкість приросту ОГ, як правило, вповільнюється після 6-го місяця життя. У даної категорії дітей відсутні неврологічні порушення при обстеженні, психологічний розвиток відповідає віку. При відсутності патологічних змін сімейна мегаленцефалія може бути підтверджена за допомогою кривих Уївера [12]. Якщо ОГ дитини знаходиться в межах норми при оцінці за кривими Уївера, відсутня необхідність проведення нейрорадіологічного обстеження. При проведенні нейровізуалізації може бути виявлене розширення супратенторіальних лікворних просторів, розширення міжпівкульної щілини. Однак у даній групі дітей незначна зовнішня гідроцефалія є нормою. Лікворні простори, як правило, нормалізуються до 3–4-річного віку. До критеріїв діагностики сімейної макроцефалії, які були вперше розроблені DeMyer, відносяться: 1) відсутність аномалій розвитку черепа, обличчя або вроджених вад розвитку, що дозволяють ідентифікувати генетичний синдром; 2) відсутність структурних змін головного мозку при нейровізуалізації; 3) наявність одного з батьків або брата чи сестри з макроцефалією, або макроцефалія, що може бути простежена у декількох поколіннях [3].
Повідомляється також, що у невеликого відсотка (приблизно 6,5 %) дітей із доброякісною сімейною макроцефалією спостерігаються незначна затримка психічного розвитку або проблеми у навчанні, тому цей стан може розглядатися як фактор ризику виникнення проблем із розвитком [3].
Макроцефалія, що супроводжується прискореним зростом окружності голови без ознак підвищення внутрішньочерепного тиску та вогнищевої неврологічної симптоматики, може виникати при непрогресуючому розширенні субарахноїдальних просторів у поєднанні з розширенням шлуночків або без нього. Такий стан у літературі отримав назву «доброякісне збільшення субарахноїдальних просторів у немовлят» (вenign enlargement of the subarachnoid spaces in infancy — BESS or BESSI), або «доброякісна гідроцефалія немовлят», або «доброякісна зовнішня гідроцефалія».
Причина цього стану на сьогодні остаточно не відома, однак, можливо, він пов’язаний із затримкою розвитку парасагітальних каналів твердої оболонки, які у маленьких дітей відповідальні за резорбцію ліквору (оскільки кількість павутинних ворсинок у цьому віці є малою). Прискорене зростання голови може тривати до віку 12–18 міс., а потім, як правило, стабілізується у вигляді макроцефалії. Нейровізуалізація при даному стані виявляє помірне збільшення бічних і третього шлуночків, розширення субарахноїдальних просторів над лобними частками та симетричне розширення міжпівкульної щілини і сільвієвої борозни. Немовлята з BESS мають підвищений ризик виникнення субдуральної гематоми, спонтанно або після незначної травми, в результаті розтягування кіркових вен. На сьогодні більшість дослідників вважають даний стан доброякісним та прогностично сприятливим [13]. У той же час отримані останнім часом дані свідчать про те, що розширення субарахноїдальних просторів, навіть за відсутності неврологічної симптоматики, може бути ранньою ознакою порушень психічного розвитку [14].
У дослідженні Shen et al. було проведено обстеження головного мозку 55 дітей віком понад 24 міс. (середній вік — 32,5 міс.) за допомогою МРТ. Метою дослідження був пошук ранніх маркерів розвитку розладів аутистичного спектра у дітей. Серед обстежених дітей 33 мали високий ризик розвитку РАС, оскільки у них були старший брат чи сестра з цим захворюванням. Діти були обстежені за допомогою МРТ тричі у віці 6–9 місяців, 12–15 та 18–24 місяців. Серед 10 дітей, у яких в подальшому розвинувся РАС, усі мали збільшений об’єм рідини у зовнішніх лікворних просторах, що виявлявся вже у віці 6–9 місяців та залишався збільшеним у 12–15 і 18–24 місяці. Крім того, діти, які згодом захворіли на РАС, мали значно більші об’єми всього головного мозку під час усіх трьох обстежень [14]. Натомість в іншому великому проспективному дослідженні окружності голови у немовлят з високим ризиком розвитку РАС протягом перших 3 років життя не було встановлено, що швидкість росту голови є предиктором розвитку РАС [15].
Стосовно поєднання макроцефалії та розладів аутистичного спектра у світовій літературі накопичено значну кількість фактів. Макроцефалія спостерігається у 15–35 % дітей із РАС. Вважається, що макроцефалія при РАС відсутня при народженні, але розвивається після 1,5–2 років. У віці близько 4 років розміри голови таких дітей зазвичай відповідають середнім у популяції [16]. У лонгітудному дослідженні встановлено, що середній загальний об’єм мозку у дітей з РАС помірно збільшується у ранньому дитинстві та згодом зменшується в дошкільному віці на відміну від дітей, які нормально розвиваються. Зменшення загального об’єму мозку при РАС у пізньому дитинстві відбувається в основному через зниження росту обсягу білої речовини півкуль мозку [17].
Під час аналізу оцінки даних інтеріктальної електроенцефалографії (ЕЕГ) у період бадьорості та сну дітей із РАС (з судомами в анамнезі або без них) G. Valvo et al. виявили епілептиформні зміни на ЕЕГ у 154 із 220 дітей. Було встановлено, що регресивні розлади спектра аутизму мають зв’язок із патологічною епілептиформною активністю у головному мозку. Виявлено вірогідну асоціацію між регресивними формами РАС та скроневою локалізацією епілептиформної активності, а також між нерегресивними РАС та локалізацією епілептиформної активності в каудальних відділах головного мозку. Крім того, в останніх дослідженнях було встановлено фенотип регресивного РАС, а саме поєднання скроневої локалізації епілептиформної активності на ЕЕГ та макроцефалії. Також при проведенні МРТ у даних дітей було виявлено зменшення товщини кори правої скроневої частки [18].
За результатами дослідження, в якому було проаналізовано 5225 пацієнтів із РАС, частота макроцефалії серед них становила 15,7 %, переважно серед пацієнтів раннього віку з низьким рівнем інтелекту. Збільшення об’єму головного мозку виявлено у 9,1 % серед 1158 МРТ-обстежень [19].
На сьогодні як основні патофізіологічні механізми збільшення розмірів мозку у дітей із РАС розглядаються такі: підвищений нейрогенез, гліогенез, збільшений синаптогенез, порушення міграції нейронів, зниження апоптозу, зниження прунінгу [20].
Важливу роль у процесах дозрівання та диференціювання мозкових клітин відіграють метаболіти поліненасичених жирних кислот — простагландини (ПГ). Дослідження показують їх вплив на процеси формування дендритних відростків, синаптичну пластичність, життєдіяльність нейронів [21–23].
Важливість простагландину E2 для забезпечення раннього нейрогенезу (11–15-та доба ембріонального розвитку) підтверджується під час проведення експериментів на мишах [24]. В експериментах при нестачі ПГ E2 були виявлені порушення розвитку багатьох мозкових структур. Хоча до кінця не відомий механізм впливу ПГ Е2 на розвиток мозкових структур, але є гіпотези, що вони впливають на WNT-сигнальний шлях розвитку головного мозку — один із внутрішньоклітинних сигнальних шляхів, що регулює ембріогенез і диференціювання клітин [25]. Wong et al. в дослідженнях показали, що ПГ E2 може виявляти модулюючиий вплив на WNT-гени, що відображається при нейруляції та проліферації нейроектодермальної трубки [25]. Медіаторні білки WNT-сигнального шляху необхідні для проліферації первинних нервових клітин кіркової міграції та синаптогенезу [27–29].
Враховуючи той факт, що WNT-сигнальний шлях є регуляторним щодо диференціювання нервових клітин, їх проліферації та міграції під час розвитку, то ПГ E2-залежне порушення регуляції цим шляхом може призвести до аномальних змін у структурі головного мозку, кіркової ектопії, формування нейронних міні-колон, що проявляються мегаленцефалією та лежать в основі аутистичних розладів [30]. Необхідне подальше вивчення механізмів впливу або взаємозв’язку між ПГ E2 та WNT-сигнальним шляхом, порушення яких проявляється у вигляді розладів аутистичного спектра.
Незважаючи на відсутність чітко встановленого зв’язку між макроцефалією та РАС, останні дослідження повідомляють про те, що пацієнти з поєднанням даних ознак часто є носіями певних мутацій. Зокрема, результати досліджень серед сиблінгів, які страждають від РАС та мають макроцефалію, показали часте виявлення мутацій гена PTEN (10q25.2) [31]. Також опубліковано кілька клінічних випадків поєднання макроцефалії з РАС у дітей з мутацією гена PTEN [32]. Останнім часом виділено окрему нозологічну одиницю — «синдром макроцефалії/аутизму», пов’язаний з геном PTEN. Мутації в гені PTEN верифікуються у 22 % дітей з поєднанням РАС, макроцефалії та епілептичних нападів. Таким чином, пошук мутації даного гена є необхідним дослідженням при поєднанні макроцефалії з РАС, особливо за наявності у пацієнта епілептичних нападів [33].
Також мутації у гені PTEN є відповідальними за розвиток синдрому Баньяна — Райлі — Рувалкаба — захворювання, що характеризується макроцефалією, високим зростом, розумовою відсталістю та різними шкірними проявами, зокрема наявністю ліпом, гемангіом, пігментних плям на шкірі, пігментаціями на статевому члені у хлопчиків, пігментацією макули [34].
Макроцефалія зустрічається також при інших захворюваннях, асоційованих з мутаціями гена PTEN. Дана група захворювань має загальну назву PTEN hamartoma tumor syndromes (PTEN-асоційовані синдроми з пухлинами і гамартомами). До них, крім синдрому Баньяна — Райлі — Рувалкаба, належать синдром Ковдена (множинні гамартоми шкіри, слизових оболонок, внутрішніх органів і залоз) і Лермітт — Дуклос (диспластична гангліоцитома мозочка) [35, 36]. Ген PTEN кодує білок-супресор росту пухлин, який діє на фермент фосфоінозитид‑3-кіназу, який бере участь в регуляції клітинного циклу, ангіогенезу і клітинної проліферації [37].
Синдром Сотоса (синдром церебрального гігантизму) — це рідкісне генетичне, у більшості випадків спорадичне (описані сімейні випадки захворювання з автосомно-домінантним типом успадкування) захворювання, що характеризується надмірним збільшенням показників соматичного розвитку протягом перших 2–3 років життя, специфічними рисами обличчя та затримкою психомовного розвитку. Поєднання даних ознак у дитини дозволяє встановити клінічний діагноз «синдром Сотоса». При синдромі Сотоса також спостерігається пренатальна макроцефалія [38].
Синдром Беквіта — Відеманна розвивається внаслідок епігенетичного дефекту 11p15, характеризується макросомією, дефектами передньої черевної стінки та макроглосією. Додатковими критеріями синдрому є насічки на мочках вух та округлі втиснення на задній поверхні потилиці, вісцеромегалія, гемігіпертрофія, неонатальна гіпоглікемія, аномалії нирок і схильність до ембріональних пухлин. Гіперглосія та відсутність характерних для синдрому Сотоса фенотипових ознак дозволяє легко диференціювати ці синдроми. За наявності значної схожості фенотипів диференціальна діагностика проводиться за допомогою молекулярно-генетичного дослідження мутацій NSD1 та 11p15 [39].
Також значна схожість спостерігається між синдромами Сотоса та Уївера. Для синдрому Уївера характерними є макроцефалія (у 83 % пацієнтів), макросомія у поєднанні з гіпертелоризмом, округлим обличчям, збільшеним кістковим віком, шкірними розтяжками, камподактилією. У пацієнта під час проведення нейровізуалізації можуть виявлятися кісти прозорої перегородки, атрофічні зміни півкуль мозку, ділянки пахігірії або гіперваскуляризації. Мутація NSD1 може виявлятися у деяких пацієнтів з фенотипом синдрому Уївера [40, 41].
Серед інших можливих причин макроцефалії необхідно враховувати такі:
— нейрошкірні захворювання. Найбільш часто макроцефалія спостерігається у пацієнтів з нейрофіброматозом 1-го типу (20–30 % дітей) [42];
— гемімегаленцефалія — може бути як ізольованою вадою розвитку головного мозку, так і відмічатися при інших генетичних захворюваннях (туберозний склероз, нейрофіброматоз 1-го типу, синдроми Кліппеля — Треноне, Протея, гіпомеланоз Іто, синдром лінійного епідермального невусу тощо) [43];
— синдром Сімпсона — Голабі — Бехмеля — характеризується макросомією від народження, макроцефалією, грубими рисами обличчя, потовщеними губами, широким ротом, макроглосією, високим піднебінням, аномаліями росту зубів, виступаючою щелепою, короткою шиєю, затримкою розвитку. Описані випадки поєднання синдрому з аномалією Арнольда — Кіарі та агенезією мозолистого тіла [1, 44];
— синдром макроцефалії — капілярної мальформації (M–CM syndrome) — характеризується наявністю макроцефалії, вродженої макросомії, гемігіпетрофії тіла, «палаючих» судиних невусів, переважно на шкірі тулуба, капілярних мальформацій на шкірі верхніх кінцівок, синдактилії або полідактилії, іноді — затримки розвитку, епілептичних нападів. На МРТ головного мозку можуть виявлятися генералізоване збільшення паренхіми, нерівномірність білої речовини, фокальні кортикальні дисплазії, полімікрогірія, вентрикуломегалія, простори Вірхова — Робіна, аномалія Арнольда — Кіарі, гемімегаленцефалія [45];
— нейросерцево-лицево-шкірні синдроми (серцево-лицево-шкірний синдром, синдроми Нунана і Костелло) — поєднують вади розвитку серцево-судинної системи (частіше — стеноз легеневої артерії), порушення росту шкіри (дифузний іхтіоз, дерматити) та волосся, кісткової системи (полі- або синдактилія), низькорослість, розумову відсталість, зміни рис обличчя, високий ризик розвитку пухлин;
— синдром Х-ламкої (фрагільної) хромосоми (синдром Мартіна — Белла). Дане захворювання є найбільш частою генетичною причиною розумової відсталості у хлопчиків. У 50 % хворих розвивається розлад аутистичного спектра. Характерними особливостями є видовжене обличчя, великі вушні раковини, макрогнотія та макроорхідизм. На МРТ виявляється збільшення хвостатого ядра, зменшення черв’яка мозочка, мигдалин, верхніх скроневих звивин, перивентрикулярні гетеротопії [46];
— синдром базальнoклітинної карциноми (синдром Горліна). Даний синдром має автосомно-домінантний тип успадкування. У пацієнтів підвищений ризик розвитку базальноклітинної карциноми в підлітковому та юнацькому віці. Особи з синдромом Горліна мають вищі показники ОГ, дисморфічне обличчя, кісти щелеп, а також долонно-підошовне заглиблення. Мутація виникає в гені pTCH1;
— метаболічна макроцефалія — виникає внаслідок накопичення продуктів метаболізму в тканині головного мозку. Окружність голови у дітей з метаболічною мегаленцефалією зазвичай в межах норми при народженні, але різко збільшується в постнатальному і навіть неонатальному періоді. Серед прикладів захворювань, що супроводжуються метаболічною макроцефалією, є лейкодистрофії (Александера, хвороба Канавана, мегаенцефалія з субкортикальними кістами), лізосомні хвороби накопичення (Тея — Сакса, мукополісахаридоз і гангліозидоз), глутарова ацидурія [1].
Як ілюстрацію наводимо клінічний випадок макроцефалії, причиною якої було генетичне захворювання (синдром Сотоса). Даний випадок ілюструє складність диференціальної діагностики макроцефалії у дітей.
Хлопчик М., 1 року, надійшов у відділення дитячої психоневрології ДУ «Інститут педіатрії, акушерства і гінекології НАМН України» зі скаргами матері на наявність у дитини затримки статокінетичного розвитку (самостійно не сідає та не стоїть), прискорене зростання окружності голови, асиметрію обличчя та різницю в об’ємі кінцівок. Дитина народилась від другої неускладненої вагітності (від першої вагітності є здоровий хлопчик). Під час пренатального обстеження TORCH-інфекцій не було виявлено. При проведенні УЗД плода діагностовано великий плід. Пологи шляхом планового кесарського розтину, маса при народженні — 4900 г (більше 2 стандартних відхилень), довжина тіла — 58 см, ОГ — 44 см, закричав відразу. За шкалою Апгар отримав 8/9 балів. Дихання після народження самостійне, судоми не відмічалися.
До 2 місяців голову не утримував, відмічалося збільшення окружності голови. За даними нейросонографії (НСГ) виявлено ознаки вентрикулодилатації 2-го ст. Було призначено дегідратаційну терапію (діакарб, аспаркам). До 5 міс. відбувалося наростання ліквородинамічних порушень за даними НСГ. У 5,5 міс. проведено МРТ голови, виявлено ознаки внутрішньої відкритої, асиметричної гідроцефалії середнього ступеня тяжкості. З 7-місячного віку приріст голови до 1 см на місяць.
У брата батька дитини народилася дитина зі збільшеною окружністю голови, синдактилією пальців на руках та ногах, екземою 30 % тіла.
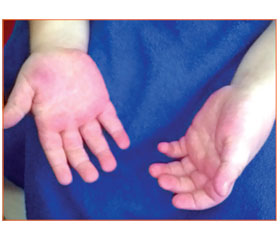
У неврологічному статусі: голова макроцефальної форми (ОГ — 52 см, розширення венозної сітки у скроневій ділянці), макросомія (маса тіла — 13 кг, зріст — 80 см), високий і широкий лоб, гіперемія у виличній ділянці, гіперемія долонь, збільшення долонь і стоп, гемігіпертрофія (праві кінцівки збільшені в об’ємі на 0,5–1,5–2,0 см, максимально виражена в проксимальних відділах, права ступня більша на 0,5 см за об’ємом та на 1,5 см за довжиною). Очні щілини D > S, сплюснута ліва половина обличчя (збільшена права), непостійний синдром Греффе. Загальна м’язова гіпотонія. Знижений тургор тканин, гіпермобільність суглобів. Голову утримує, сидить, але сам не сідає, опір на ноги слабкий. Рівень психомовного розвитку знижений. Під час обстеження в загальному аналізі крові відхилень не виявлено; у біохімічному аналізі крові: гіпербілірубінемія — 26,0 мкмоль/л (норма — до 20,5 мкмоль/л); загальний аналіз сечі — без відхилень; НСГ — ознаки вентрикулодилатації; електрокардіографія (ЕКГ) — зниження провідності по правій ніжці пучка Гіса; ехокардіографія — відкрите овальне вікно; фіброгастродуоденоскопія — недостатність кардії, рефлюкс-езофагіт; УЗД-ознаки гіперплазії тимуса. Враховуючи наявність у дитини характерного фенотипу обличчя, макроцефалії, макросомії, загальної м’язової гіпотонії, дисплазії сполучної тканини, гемігіпертрофії, дані об’єктивного обстеження, встановлений діагноз: синдром Сотоса (підтвердженням діагнозу є обтяжений спадковий анамнез по батьківській лінії — дитина з макросомією, макроцефалією, полісиндактилією, екземою).
При повторній госпіталізації дитини в 1 рік 2 міс. відмічена затримка психомовленнєвого розвитку (не говорить, не виконує інструкцій для даного віку) та статокінетичного розвитку (самостійно не сідає та не ходить). Зберігаються прискорений ріст окружності голови та різниця в об’ємі кінцівок. Макроцефалія (ОГ — 56 см, окружність грудної клітки (ОГК) — 51,5 см, велике тім’ячко (ВТ) — 3 × 4 см, не напружене), макросомія, асиметрія мімічної іннервації, D > S, очні щілини D > S. Збільшені в об’ємі долоні та стопи. Праві кінцівки більші в об’ємі на 1,5–2 см (спостерігається зростання різниці в об’ємі), в довжині близько 0,5 см, гіпермобільність суглобів, слабкість опори. У загальному аналізі крові, сечі та біохімічному аналізі крові патологічних змін не виявлено. При проведенні МРТ виявлено аномалію Арнольда — Кіарі І, порушення ліквородинаміки на рівні великого потиличного отвору. УЗД органів черевної порожнини — без патологічних відхилень. НСГ — наявність вентрикулодилатації 2-го ст. ЕКГ — неповна блокада правої ніжки пучка Гіса. При цитогенетичному дослідженні встановлено нормальний чоловічий каріотип (46ХУ).
При зверненні в 1 рік 8 міс. у дитини відмічено затримку статокінетичного розвитку (самостійно не стоїть, не ходить) та психомовленнєвого розвитку (не говорить, інструкції не виконує). Прискорений об’єм росту голови (за 4 міс. приріст ОГ з 56 до 57 см), зростання різниці об’єму кінцівок не виявлено, макросомія, асиметрія при оцінці черепно-мозкових нервів D > S та очних щілин D > S. Збільшені в об’ємі долоні та стопи. Праві кінцівки більші в об’ємі на 1,5–2 см, в довжині близько 0,5 см, гіпермобільність суглобів, слабкість опори. У загальному аналізі крові, сечі та біохімічному аналізі крові патологічних змін не виявлено. УЗД органів черевної порожнини — без патологічних відхилень. НСГ — наявність ліквородинамічних порушень. ЕКГ — неповна блокада правої ніжки пучка Гіса. Висновок нейрохірурга: гідроцефалія в стадії компенсації з ознаками часткової оклюзії водопроводу Моро, мальформація Кіарі І. Висновок кардіолога: даних за ваду розвитку не виявлено, відкрите овальне вікно.
При надходженні в 2 роки дитина говорить декілька слів, виконує прості інструкції, самостійно не сідає та не ходить. Відмічається різниця в об’ємі кінцівок, макросомія та макроцефалія. У неврологічному статусі: ОГ — 59 см, ОГК — 60 см, ВТ закрите, маса тіла — 22 кг. Відмічена асиметрія обличчя D > S, тонус м’язів і тургор тканин знижені. Слух і жування, ковтання не порушені. Координація рухів не порушена. Об’єм правої кінцівки більше на 1–2 см, ліва нога коротша на 2–3 см. Самостійно не стоїть та не ходить. Вимовляє 3–4 слова, виконання інструкцій на рівні дитини 1 року, розширення судинної сітки на шкірі долонь і стоп. Висновок МРТ: з найбільшою ймовірністю має місце правостороння гемімегаленцефалія; МР-ознаки аномалії Арнольда — Кіарі І. Порівняно з попереднім МРТ відмічається зменшення ступеня розширення лівого бокового шлуночка. Висновок нейрохірурга: макроцефалія, гідроцефалія в стадії компенсації, мальформація Арнольда — Кіарі І. Ознак внутрішньочерепної гіпертензії не виявлено. Висновок генетика: з найбільшою ймовірністю синдром Сотоса. Лабораторних відхилень не виявлено.
У віці 6 років у дитини спостерігається відставання у психічному розвитку (легка розумова відсталість), загальне недорозвинення мовлення 2-го рівня, легка тулубна атаксія, макросомія (рис. 1А), макроцефалія, гемігіпертрофія (стабільна), розширена судинна сітка на долонях і стопах (рис. 1Б, 2).
Висновки
У даній публікації ми спробували коротко проаналізувати основні причини макроцефалії у дітей. Незважаючи на значні досягнення молекулярної генетики, які останніми десятиріччями дозволили встановити велику кількість генів, відповідальних за розвиток макроцефалії, патогенез даного стану залишається до кінця не відомим. У більшості випадків диференціальній діагностиці макроцефалії сприяє клінічний огляд пацієнта та ретельний збір анамнезу. Візуальний і неврологічний огляд допомагають запідозрити значну кількість генетичних захворювань, що супроводжуються макроцефалією, у тому числі факоматози, синдром Х-ламкої хромосоми, синдроми, що супроводжуються макросомією. Методи нейровізуалізації, зокрема МРТ головного мозку, можуть допомогти виключити метаболічні захворювання, пухлини, вроджені вади розвитку, гідроцефалію. Правильна диференціальна діагностика макроцефалії надзвичайно важлива, оскільки дозволяє розпочати адекватні лікувально-реабілітаційні заходи. Слід також пам’ятати, що даний стан може розцінюватись як фактор ризику розвитку розладу аутистичного спектра, особливо за наявності структурних змін мозку на МРТ, або обтяженого сімейного анамнезу (наявності сибілінгів, які страждають від РАС).
Список литературы
1. Williams C., Dagli A., Battaglia A. Genetic disorders associated with macrocephaly // Am. J. Med. Genet. Part. A. — 2008. — 146A. — Р. 2023-2037.
2. Seal A. Fifteen-minute consultation on the infant with a large head // Arch. Dis. Child Educ. Pract. Ed. — 2013 Aug. — 98(4). — Р. 122-5.
3. DeMyer W. Megalencephaly: Types, clinical syndromes, and management // Pediatr. Neurol. — 1986. — 2. — Р. 321-328.
4. Bodensteiner J., Chung E. Macrocrania and megalencephaly in the neonate // Semin. Neurol. — 1993. — 13. — Р. 84-91.
5. Клінічний протокол медичного догляду за здоровою дитиною віком до 3 років. Затверджено МОЗ України від 20.03.2008, № 149.
6. DiLiberti J.H. Inherited macrocephaly-hamartoma syndromes // Am. J. Med. Genet. — 1998. — 79. — Р. 284-290.
7. Fenichel G. Disorders of cranial volume and shape // Clinical. Pediatric. Neurology: A Signs and Symptoms Approach. — 5th. — Philadelphia: Elsevier Saunders, 2005. — 353 р.
8. Kurmanavicius J., Wright E., Royston P. et al. Fetal ultrasound biometry: 1. Head reference values // Br. J. Obstet. Gynaecol. — 1999. — 106. — Р. 126.
9. Biran-Gol Y., Malinger G., Cohen H. et al. Developmental outcome of isolated fetal macrocephaly // Ultrasound Obstet. Gynecol. — 2010. — 36. — Р. 147.
10. Gleeson J., Dobyns W., Plawner L. et al. Congenital structural defects // Pediatric Neurology Principles and Practice. — 4th ed. / Swaiman K., Ashwal S., Ferriero D.M. — Philadelphia: Mosby Elsevier, 2006. — 399 р.
11. Lorber J., Priestley B. Children with large heads: A practical approach to diagnosis in 557 children, with special reference to 109 children with megalencephaly // Dev. Med. Child Neurol. — 1981. — 23. — Р. 494-504.
12. Weaver D., Christian J. Familial variation of head size and adjustment for parental head circumference // J. Pediatr. — 1980. — 96. — Р. 990.
13. Vertinsky A.T., Barnes P.D. Macrocephaly, increased intracranial pressure, and hydrocephalus in the infant and young child // Top. Magn. Reson. Imaging. — 2007 Feb. — № 18(1). — Р. 31-51.
14. Shen M., Nordahl C., Young G.S. et al. Early brain enlargement and elevated extra-axial fluid in infants who develop autism spectrum disorder // Brain. — 2013 Sep. — № 136(Pt. 9). — Р. 2825-35.
15. Zwaigenbaum L., Young G., Stone W. et al. Early head growth in infants at risk of autism: A Baby Siblings Research Consortium Study // J. Am. Acad. Child Adolesc. Psychiatry. — 2014. — № 53. — Р. 1053-1062.
16. Stanfield A., McIntosh A. et al. Towards a neuroanatomy of autism: a systematic review and meta-analysis of structural magnetic resonance imaging studies // Eur. Psychiatry. — 2008. — № 23(4). — Р. 289-99.
17. Lange N., Travers B.G., Bigler E.D. et al. Longitudinal volumetric brain changes in autism spectrum disorder ages 6-35 years // Autism Res. — 2014; Autism Res. — 2015 Feb. — № 8(1). — Р. 82-93.
18. Valvo G. еt al. Temporal lobe connects regression and macrocephaly to autism spectrum disorders // Eur. Child Adolesc. Psychiatry. — 2015 July. — Р. 1-9.
19. Sacco R., Gabriele S., Persico A.M. Head circumference and brain size in autism spectrum disorder: A systematic review and meta-analysis // Psychiatry Res. — 2015 Nov 30. — № 234(2). — Р. 239-51.
20. Saskia J., Palmen M.C. Structural Brain Abnormalities in Autism: Neuroimaging and Neuropathology Studies, 2005. — 28 р.
21. Burks S.R., Wright C.L., McCarthy M.M. Exploration of prostanoid receptor subtype regulating estradiol and prostaglandin E2 induction of spinophilin in developing preoptic area neurons // Neuroscience. — 2007. — № 146(3). — Р. 1117-27.
22. Koch H., Huh S.E., Elsen F.P. et al. Prostaglandin E2-induced synaptic plasticity in neocortical networks of organotypic slice cultures // J. Neurosci. — 2010. — № 30(35). — Р. 11678-87.
23. Jiang J., Ganesh T., Du Y. et al. Neuroprotection by selective allosteric potentiators of the EP2 prostaglandin receptor // Proc. Natl. Acad. Sci. USA. — 2010. — № 107(5). — Р. 2307-12.
24. Tamiji J., Crawford D.A. The neurobiology of lipid metabolism in autism spectrum disorders // Neurosign. — 2010. — № 18. — Р. 98-112.
25. Evans T. Fishing for a WNT-PGE2 link: beta-catenin is caught in the stem cell net-work // Cell Stem Cell. — 2009. — № 4(4). — Р. 280-2.
26. Wong C., Li H., Crawford D. The role of prostaglandin E2 signaling pathway in neuroectodermal stem cell function. Poster# 151.08/V6. Society for Neuroscience — Annual Meeting; Sunday, 13 Nov 2011. — Washington, DC, 2011.
27. Zhou C., Zhao C., Pleasure S. Wnt signaling mutants have decreased dentate granule cell production and radial glial scaffolding abnormalities // J. Neurosci. — 2004. — № 24(1). — Р. 121-6.
28. Zhou C.J., Borello U., Rubenstein J., Pleasure S. Neuronal production and precursor proliferation defects in the neocortex of mice with loss of function in the canonical Wnt signaling pathway // Neuroscience. — 2006. — № 142(4). — Р. 1119-31.
29. Sahores M., Salinas P. Activity-mediated synapse formation a role for Wnt-Fz signaling // Curr. Top. Dev. Biol. — 2011. — № 97. — Р. 119-36.
30. Buechling T., Boutros M. Wnt signaling signaling at and above the receptor level // Curr. Top. Dev. Biol. — 2011. — № 97. — Р. 21-53.
31. Spence S., Kono N., Geschwind D., Cantor R. A QTL Analysis of Macrocephaly in Autism Families. American Society of Human Genetics Annual Meeting. — Salt Lake City, UT., 2005.
32. Herman G., Butter E., Enrile B. et al. Increasing knowledge of PTEN germline mutations: Two additional patients with autism and macrocephaly // Am. J. Med. Genet. — 2007. — Part A(143A). — Р. 589-593.
33. Marchese M., Conti V., Valvo G. et al. Autism-epilepsy phenotype with macrocephaly suggests PTEN, but not GLIALCAM, genetic screening // BMC Med. Genet. — 2014 Feb 27. — № 15. — Р. 26. — Doi: 10.1186/1471-2350-15-26.
34. DiLiberti J. Inherited macrocephaly-hamartoma syndromes // Am. J. Med. Genet. — 1998. — № 79. — Р. 284-290.
35. Robinson S., Cohen A. Cowden disease and Lhermitte-Duclos disease: an update. Case report and review of the literature // Neurosurg. Focus. — 2006 Jan 15. — № 20(1). — E6.
36. Perez-Nunez A., Lagares A. et al. Lhermitte-Duclos disease and Cowden disease: Clinical and genetic study in five patients with Lhermitte-Duclos disease and literature review // Acta Neurochir. (Wien). — 2004. — № 146. — Р. 679-690.
37. Hay N. The Akt-mTOR tango and its relevance to cancer // Cancer Cell. — 2005. — № 8. — Р. 179-183.
38. Tatton-Brown K., Douglas J. Genotype-phenotype associations in Sotos syndrome: an analysis of 266 individuals with NSD1 aberrations // Am. J. Hum. Genet. — 2005. — № 77. — Р. 193-204.
39. Gosden R., Trasler J., Lucifero D., Faddy M. Rare congenital disorders, imprinted genes, and assisted reproductive technology // Lancet. — 2003 Jun 7. — № 361(9373). — Р. 1975-7.
40. Opitz J., Weaver D., Reynolds J. The syndromes of Sotos and Weaver: Reports and review // Am. J. Med. Genet. — 1998. — № 79. — Р. 294-304.
41. Douglas J., Hanks S., Temple I. et al. NSD1 mutations are themajor cause of Sotos syndrome and occur in some cases of Weaver syndrome but are rare in other overgrowth phenotypes // Am. J. Hum. Genet. — 2003. — № 72. — Р. 132-143.
42. Szudek J., Birch P., Friedman J. Growth charts for young children with neurofibromatosis 1 (NF1) // Am. J. Med. Genet. — 2000. — № 92. — Р. 224-228.
43. Sasaki M., Hashimoto T., Furushima W. et al. Clinical aspects of hemimegalencephaly by means of a nationwide survey // J. Child Neurol. — 2005. — Р. 337-341.
44. Young E., Wishnow R., Nigro M. Expanding the clinical picture of Simpson-Golabi-Behmel syndrome // Pediatr. Neurol. — 2006. — № 34. — Р. 139-142.
45. Mirzaa G.M., Conway R.L., Gripp K.W. et al. Megalencephaly-capillary malformation (MCAP) and megalencephaly-polydactyly-polymicrogyria-hydrocephalus (MPPH) syndromes: two closely related disorders of brain overgrowth and abnormal brain and body morphogenesis // American Journal of Medical Genetics. — 2012 Feb. — № 158A(2). — Р. 269-91.
46. Mash Eric J. Intellectual Disability (Intellectual Developmental Disorder). — 6th ed. — Boston, Massachusetts: Cengage Learning, 2015.

/94.jpg)