
Международный неврологический журнал 7 (85) 2016
Вернуться к номеру
Випадок нейрофіброматозу 2-го типу з множинними пухлинами центральної нервової системи у вагітної жінки
Авторы: Костюченко А.В., Титаренко Н.В.
Вінницький національний медичний університет імені М.І. Пирогова, м. Вінниця, Україна
Рубрики: Неврология
Разделы: Справочник специалиста
Версия для печати
Резюме
Наведено випадок власного клінічного спостереження нейрофіброматозу 2-го типу з множинними пухлинами центральної нервової системи у вагітної жінки.
Приведен случай собственного клинического наблюдения нейрофиброматоза 2-го типа с множественными опухолями центральной нервной системы у беременной женщины.
A case of clinical observation of neurofibromatosis type II with multiple tumors of central nervous system in a pregnant woman is described.
Ключевые слова
нейрофіброматоз; множинні пухлини центральної нервової системи; вагітність
нейрофиброматоз; множественные опухоли центральной нервной системы; беременность
neurofibromatosis; multiple tumors of central nervous system; pregnancy
Статтю опубліковано на с. 60-64
Вступ
Нейрофіброматоз 2-го типу (НФ2) — це прогресуюче інвалідизуюче моногенне автосомно-домінантне захворювання, що характеризується утворенням двобічних вестибулярних шваном, множинних пухлин центральної та периферичної нервової системи, ранньою катарактою [7, 9, 13]. Це захворювання відноситься до групи факоматозiв (від грец. phakos — пляма, mitosis — пухлина) — вродженої дисплазії ембріоекто- та ендодерми, що характеризується ураженням шкіри, центральної та периферичної нервової системи, рідше — кiсткової системи, очей, інколи — внутрiшнiх органiв.
За даними D.G. Evans (2009), поширеність НФ2 (спочатку оцінювалася як 1 на 200 000 населення) становить близько 1 на 60 000 [21]. J. Antinheimo et al. (2000) повідомляли про більш низьку частоту цього захворювання — 1 на 87 410 населення [16].
Центральний НФ виникає внаслідок точкової мутації НФ2 гена, який міститься в 22-й хромосомі (22q12.2) [18]. Цей ген кодує синтез супресора пухлинного росту — білка мерліну або шваноміну, який забезпечує динамічний контроль клітинного росту. Повна інактивація цього гена викликає аномальний ріст пухлин. Для НФ2 типовою є висока частота спонтанних мутацій, в результаті чого 50 % клінічних випадків є спорадичними. Захворювання характеризуються 100% пенетрантністю та широкою фенотиповою варіабельністю.
Патогномонічним для НФ2 є наявність двобічних вестибулярних неврином, а також шваном інших черепно-мозкових, спінальних і шкірних нервів при мінімальних шкірних та екстраневральних симптомах. При НФ2, окрім вестибулярних шваном, патологічним субстратом захворювання є також об’ємні утворення різної гістологічної належності й локалізації: черепно-мозкові та спінальні менінгіоми, гліоми, нейрофіброми, епендімоми, астроцитоми [18]. Згідно з дiагностичними критеріями Національного інституту здоров’я США (National Institutes of Health — NIH) [14], НФ2 може бути встановлений за наявностi двох чи бiльше наступних ознак:
1. Двобічні новоутворення 8-го черепного нерва, візуалізовані на комп’ютерній (КТ) або магнітно-резонансній томографії (МРТ).
2. Наявність родичів 1-го порядку з НФ2 та одностороннім новоутворенням 8-го нерва або двох із наведених нижче захворювань:
— гліома;
— менінгіома;
— шванома;
— нейрофіброма;
— ювенільне заднє підкапсулярне чечевицеподібне затемнення кришталика (рання катаракта).
Наведемо власне клінічне спостереження.
Опис випадку
Вагітна С., 16 років, надійшла в клініку зі скаргами на біль у попереку, зниження слуху на праве вухо, виражену хиткість при ходьбі до падіння, різку слабкість та терпкість лівої ноги, помірну слабкість правої ноги, періодичну затримку сечовипускання, запори. На 3-тю добу перебування в стаціонарі — парез лівої ноги, терпкість лівої ноги, гіперрефлексія з обох ніг, сильніше ліворуч; на 5-ту добу — неможливість самостійної ходьби, порушення функції тазових органів за типом затримки сечі та калу, значний больовий синдром уздовж хребта, більше в грудному відділі.
Діагноз: нейрофіброматоз 2-го типу, множинні пошкодження головного мозку, нейрофіброма мостомозочкового кута справа, гліома спинного мозку на рівні Т8-Т9 сегмента, нейрофіброма Т11 корінця справа. Нижня параплегія, розлади чутливості, тазових органів. Вагітність 32–33 тижні в 16 років.
Анамнез захворювання. Захворювання виявлено у віці 13 років. Відзначений нестійкий горизонтальний ністагм при погляді вліво, екзофтальм зліва. Періостальні та сухожилкові рефлекси з ніг підвищені, D < S. Гострота зору зліва знижена до 0,4. Консультація спеціалістів: невролог — правобічний нижній монопарез, окуліст — субатрофія зорового нерва зліва. Змін з боку шкірних покривів і підшкірної клітковини немає. Серед родичів І ступеня спорідненості у матері було діагностовано НФ.
Від генетичного дослідження вагітна відмовилася.
З боку внутрішніх органів при фізикальному обстеженні патологічних змін виявлено не було.
Неврологічний статус на 5-й день перебування у стаціонарі. Менінгеальні знаки відсутні. Очні щілини та зіниці D = S, фотореакція задовільна. Об’єм рухів очима повний, диплопії немає, горизонтальний мілкорозмашистий ністагм при погляді вбоки. Тригемінальні точки не болючі. Обличчя асиметричне, язик по середній лінії. Помірне зниження слуху на праве вухо. Фонація, ковтання не порушені. Сила м’язів у м’язах нижніх кінцівок знижена більше справа (сила м’язів лівої ноги 4 бали, правої — 3 бали), м’язовий тонус кінцівок знижений, більше нижніх, більше правої ноги. Відмічається помірна гіпотрофія м’язів правої ноги. Позитивні симптоми натягу справа. Вібраційна чутливість на нижніх кінцівках знижена, більше справа. Поверхнева чутливість знижена до рівня Тh10. Сухожилкові та періостальні рефлекси з рук D = S, середньої жвавості, колінні D < S, середньої жвавості, ахіллові S < D. Патологічні стопні рефлекси відсутні, позитивні субкортикальні рефлекси. Самостійна ходьба неможлива за рахунок больового синдрому, нижнього парапарезу та атаксії, координаторні проби верхніми кінцівками виконує задовільно, нижніми — не виконує за рахунок больового синдрому та парезу. Функція тазових органів порушена за типом затримки сечі та калу, сечовипускання по постійному катетеру.
МРТ головного мозку (серпень 2013 р.): у латеральних відділах правої гемісфери мозочка вогнище гіперінтенсивного МР-сигналу на Т2ВІ, гіпоінтенсивного МР-сигналу на Т1ВІ, лінейної форми, розмірами 1,7 × 0,4 см. Об’ємне утворення правого мостомозочкового кута.
МРТ шийного відділу хребта (серпень 2013 р.): на рівні краніовертебрального переходу справа вогнище з чіткими контурами, овальної форми, ізоінтенсивного МР-сигналу на Т2ВІ та Т1ВІ, інтенсивно та однорідно накопичує контрастну речовину, розмірами 1 × 1,2 × 1,3 см, обумовлено позамозковою пухлиною. Аналогічне вогнище на рівні С1-С2 справа, інтенсивно та однорідно накопичує конт–растну речовину, розмірами 1,5 × 1,6 × 1,6 см, також обумовлене позамозковою пухлиною.
МРТ головного мозку (жовтень 2013 р.): у лівій скроневій ділянці на рівні підкоркових ядер і паравентрикулярно на рівні задніх рогів бічних шлуночків вогнища гліозного ураження. Справа (проекція міжхребцевого отвору С1-С2) солідне новоутворення розміром до 26,9 × 13,3 × 28,1 мм, з розширенням і деформацією міжхребцевого отвору та помірно вираженою деформацією лікворного простору.
МРТ головного мозку (березень 2016 р.): зберігається об’ємне утворення правого мостомозочкового кута та міжхребцевого отвору С1-С2 без збільшення розмірів. Щілеподібне кістозне утворення в правій півкулі мозочка і гліозний фокус субкортикально в лівій островковій долі без змін.
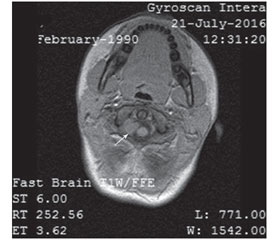
МРТ головного мозку (липень 2016 р.): вогнищеве ураження субкортикальної білої речовини в лівій острівцевій та задньолобній долі та лобноскроневій долі справа, правій півкулі мозочка, об’ємне утворення правого мостомозочкового кута (рис. 1) та міжхребцевого отвору С1-С2 (рис. 2) без змін в динаміці.
/60-64/62-1.jpg)
МРТ спинного мозку з внутрішньовенним контрастуванням (гадоліній) (липень 2016 р.): інтрадурально додаткові об’ємні утворення на рівні Тh8-Тh9 та Тh11 сегментів: у Тh8-Тh9 сегменті спинний мозок веретеноподібно розширений 14 × 12 мм за рахунок вузла в товщі розміром 5 × 9 × 22 мм, що не накопичує контраст. У Тh11 сегменті справа (екстрамедулярно в проекції корінця) визначається утворення розміром 7 × 5 × 19 мм, що не накопичує контраст. Висновок: гліома спинного мозку на рівні Тhы8-Тh9 сегмента, нейрофіброма Тh11 корінця справа (рис. 3).
/60-64/62-2.jpg)
Обстеження. Загальний аналіз крові: Er — 3,7 Т/л, Hb — 111 г/л, КП — 0,9, Тr — 236,8 Т/л, Le — 6,4 Г/л, п — 11 %, с — 68 %, е — 1 %, л — 17 %, м — 3 %, швидкість осідання еритроцитів — 43 мм/год. Біохімічний аналіз крові, загальний аналіз сечі — в межах норми, антитіла до ВІЛ виявлені не були. На електрокардіограмі патологічні зміни відсутні.
Лікування: дексаметазон, фуросемід, парацетамол, диклофенак, налбуфін.
Вагітна була розроджена шляхом кесаревого розтину, без ускладнень.
Обговорення
Нейрофіброматоз є найбільш поширеним із 54 факоматозів, описаних у каталозi спадкових захворювань McKusik [3]. На сьогодні немає єдиної класифікації НФ. Ряд авторів виділяють 7 форм цього захворювання, але найбільшого поширення набув поділ НФ на тип 1 (НФ1) та тип 2 (НФ2) [5]. Молекулярно-генетичні дослідження (результати опубліковані в 1985 і 1987 р.) виявили принципові відмінності в етіопатогенезі НФ1 і НФ2 та довели, що це абсолютно різні автосомно-домінантні генетичні захворювання [12]. Їх локуси знаходяться відповідно на хромосомах 17q11.2 та 22q12.2 [18]. Розташовані тут гени кодують синтез супресорів пухлинного росту (білків нейрофіброміну та мерліну відповідно), які забезпечують динамічний контроль клітинного росту. Повна інактивація цих генів викликає аномальний ріст пухлин. При цьому НФ2 обмежений, як правило, нервовою системою, тоді як НФ1 є системним розладом.
Класичний периферичний НФ, або хвороба Реклінгаузена, або НФ 1-го типу (НФ1), становить до 95 % випадків серед усіх хворих на нейрофіброматоз [1]. Поширеність НФ1 — 1 випадок на 5 тис. населення [4]. Частота спонтанної мутації гена NF1 у 17q-хромосомі (17q11.2) оцінюється як 1 × 10–4, що є однією з найбільших у людини [6]. Ризик успадкування дитиною даної патології при наявності НФ1 у одного з батьків дорівнює 50 %, у обох — 66,7 % [8].
НФ2 зустрічається набагато рідше, згідно з останніми даними, його частота становить близько 1 на 60 000 [21]. J. Antinheimo et al. (2000) повідомляли про ще більш низьку частоту цього захворювання — 1 на 87 410 населення [16]. Подібний клінічний випадок у неврологічній практиці можна зустріти нечасто.
Для НФ2 також є типовою висока частота спонтанних мутацій, в результаті чого 50 % клінічних випадків є спорадичними. В описаному нами клінічному спостереженні пацієнтка мала обтяжений спадковий анамнез щодо НФ2. Незважаючи на те, що від генетичного дослідження вона відмовилася, захворювання було діагностовано на підставі клінічних ознак згідно з NIH-дiагностичними критеріями [14], а саме: наявності родичів 1-го порядку з НФ2 (матір пацієнтки) та одностороннього новоутворення 8-го нерва. D.G. Evans et al. (1995) встановили, що пацієнти, які успадкували патологічний ген НФ2 від матері, мають більш тяжкий перебіг захворювання та ранній початок НФ2 [7].
НФ 2-го типу (стара назва — центральний нейрофіброматоз), який характеризується розвитком доброякісних новоутворень у центральній нервовій системі, являє особливий інтерес для неврологів [15]. Особливостями НФ2 є рання вікова клінічна маніфестація та наявність більше трьох пухлин у одного хворого; менша клінічна гетерогенність порівняно з нейрофіброматозом 1-го типу. Виникаючі при НФ2 пухлини є доброякісними, але більш біологічно агресивними, ніж при НФ1.
При підозрі на НФ2 стандартом обстеження для верифікації діагнозу є МРТ головного мозку із зрізами 3 мм через внутрішні слухові канали в сагітальній та аксіальній проекціях із контрастуванням і без нього. Причому МРТ не можна замінити КТ [2]. Патогномонічними для НФ2 є виявлення на МР-томограмах двобічних вестибулярних шваном. Їх виявляють більше ніж у 90 % дорослих пацієнтів.
НФ обох типів скоріш за все не впливає на фертильність [19], тому хворі з цим захворюванням можуть бути потенційними пацієнтками акушерських стаціонарів. Оскільки середній вік клінічної маніфестації НФ2, як правило, становить від 18 до 24 років, а середній вік на момент встановлення діагнозу — приблизно 28 років, то у пацієнток акушерських стаціонарів найчастіше мають місце початкові симптоми захворювання, обумовлені невриномою слухового нерва (44,4 %), іншими пухлинами центральної нервової системи (22,2 %), пухлинами шкіри (12,7 %), а також очні прояви, включаючи катаракту та гамартоми сітківки ока (12,7 %). За даними A. Drouet et al. (2014), офтальмологічні прояви є найбільш частою початковою ознакою НФ2 у дитячому віці [20].
Вагітність часто провокує початок захворювання, збільшення кількості та розмірів шкірних нейрофібром, а також прискорення росту пухлин центральної нервової системи за рахунок наявності в них чутливих до гормонів рецепторів [11]. Пухлини, що розвиваються в центральній нервовій системі, як правило, втягують у процес чутливі нерви, які також мають високу концентрацію рецепторів до жіночого гормона естрогену [10]. Подібний випадок був описаний у породіллі з НФ2, у якої зареєстровано прогресування симптомів, імовірно, через збільшення розміру пухлини лівого плечового сплетіння [13]. Таким чином, потенціальне швидке збільшення кількості та розмірів пухлин центральної нервової системи під час вагітності необхідно враховувати у пацієнток як із НФ1, так і з НФ2. У нашому випадку у пацієнтки впродовж 5 діб відбулося прогресування неврологічного дефіциту від помірного парезу лівої ноги до нижньої параплегії, розладів тазових органів і чутливості знизу до рівня Тh10, що було обумовлено компресією спинного мозку гліомою на рівні Тh8-Тh9 сегмента (рис. 2). Виражений неврологічний дефіцит є основною ознакою НФ2 [9].
Висновки
Нейрофіброматоз є складною проблемою сучасної медицини, що обумовлено значною фенотиповою та клінічною варіативністю захворювання, складністю його діагностики та лікування, зниженням якості життя хворих. Вагітність часто провокує початок захворювання, збільшення кількості та розмірів шкірних нейрофібром, а також прискорення росту пухлин центральної нервової системи за рахунок наявності в них чутливих до гормонів рецепторів. Ведення вагітних із нейрофіброматозом вимагає координованого міждисциплінарного підходу при виборі акушерської тактики, анестезіологічного забезпечення, подальшого спостереження та лікування.
Конфлікт інтересів. Автори заявляють про відсутність конфлікту інтересів при підготуванні даної статті.
Список литературы
1. Квасніцький М.В. Нейрофібрматоз‑1 (огляд літератури) / М.В. Квасніцький // Український нейрохірургічний журнал. — 2001. — № 4. — С. 13-23.
2. Квасніцький М.В. Нейрофіброматоз‑2 (огляд літератури) / М.В. Квасніцький // Український нейрохірургічний журнал. — 2002. — № 2. — С. 14-21.
3. Короленко В.В. Випадок нейрофіброматозу І типу з ураженням головного мозку / В.В. Короленко, В.Г. Коляденко // Дерматологія. — 2009. — № 1. — С. 34-35.
4. Цимбалюк В.І. Діагностика та диференційоване лікування нейрофіброматозу 1-го типу / В.І. Цимбалюк, М.В. Квасніцький // Український медичний часопис. — 2003. — № 4. — С. 97-104.
5. Цимбалюк В.І. Спроба класифікації нейрофіброматозу / В.І. Цимбалюк, М.В. Квасніцький // Вісник Сумського державного університету. Серія «Медицина». — 2003. — № 9(55). — С. 100-107.
6. A genetic study of von Recklinghausen neurofibromatosis in south east Wales. I. Prevalence, fitness, mutation rate, and effect of parental transmission on severity / S.M. Huson, D.A. Compston, P. Clark, P.S. Harper // J. Med. Genet. — 1989. — Vol. 26, № 11. — P. 704-711.
7. Evans D.G. Neurofibromatosis type 2 (NF2): a clinical and molecular review / D.G. Evans // Orphanet. J. Rare Dis. — 2009. — Vol. 4. — P. 16.
8. http://www. neurofibromatos1.narod.ru/ index.htm.
9. Lloyd S.K. Neurofibromatosis type 2 (NF2): diagnosis and management / S.K. Lloyd, D.G. Evans // Handb. Clin. Neurol. — 2013. — Vol. 115. — P. 957-967.
10. Martuza R.L. Specific estradiol binding in schwannomas, meningiomas, and neurofibromas / R.L. Martuza, D.T. Mac–Laughlin, R.G. Ojemann // Neurosurgery. — 1981. — Vol. 9, № 6. — P. 665-671.
11. McLaughlin M.E. Progesterone receptor expression in neurofibromas / M.E. McLaughlin, T. Jacks // Cancer Res. — 2003. — Vol. 63, № 4. — P. 752-755.
12. Neurofibromatoses: part 1 — diagnosis and differential diagnosis / L.O. Rodrigues, P.B. Batista, E.M. Goloni-Bertollo [et al.] // Arq. Neuropsiquiatr. — 2014. — Vol. 72, № 3. — P. 241-250.
13. Neurofibromatosis 2 (NF2): clinical characteristics of 63 affected individuals and clinical evidence for heterogeneity / D.M. Parry, R. Eldridge, M.I. Kaiser-Kupfer [et al.] // Am. J. Med. — 1994. — Vol. 52, № 4. — P. 450-461.
14. Neurofibromatosis. Conference statement. National Institutes of Health Consensus Development Conference // Arch. Neurol. — 1988. — Vol. 45, № 5. — P. 575-578.
15. Nowak C.B. The phakomatoses: dermatologic clues to neurologic anomalies / C.B. Nowak // Semin. Pediatr. Neurol. — 2007. — Vol. 14, № 3. — P. 140-149.
16. Population-based analysis of sporadic and type 2 neurofibromatosis-associated meningiomas and schwannomasis / J. Antinheimo, R. Sankila, O. Carpén [et al.] // J. Neurology. — 2000. — Vol. 54, № 1. — P. 71-76.
17. Riccardi V.M. Neurofibromatosis: clinical heterogeneity / V.M. Riccardi // Curr. Problem Cancer. — 1992. — Vol. 7, № 2. — P. 1-34.
18. Ruggieri M. Diagnosis, Management, and New Therapeutic Options in Childhood Neurofibromatosis Type 2 and Related Forms / M. Ruggieri, A.D. Praticò, D.G. Evans // Semin. Pediatr. Neurol. — 2015. — Vol. 22, № 4. — P. 240-258.
19. Sakai T. A parturient with neurofibromatosis type 2: anesthetic and obstetric considerations for delivery / T. Sakai, M.C. Vallejo, K.T. Shannon // Int. J. Obstet. Anesth. — 2005. — Vol. 14, № 4. — P. 332-335.
20. Type 2 neurofibromatosis: intergenerational differences in genetic and clinical expression / A. Drouet, F. Le Moigne, D. Salamé [et al.] // Arch. Pediatr. — 2014. — Vol. 21, № 11. — P. 1233-1240.
21. Variation of expression of the gene for type 2 neurofibromatosis: absence of a gender effect on vestibular schwannomas, but confirmation of a preponderance of meningiomas in females / D.G. Evans, V. Blair, T. Strachan [et al.] // J. Laryngol. Otol. — 1995. — Vol. 109, № 9. — P. 830-835.