
Международный эндокринологический журнал Том 13, №4, 2017
Вернуться к номеру
Чоловічий гіпогонадизм (Частина 1)
Авторы: Лучицький Є.В.(1, 2), Лучицький В.Є.(1), Тронько М.Д.(1, 2)
(1) — ДУ «Інститут ендокринології та обміну речовин ім. В.П. Комісаренка НАМН України», м. Київ, Україна
(2) — Національна академія післядипломної освіти імені П.Л. Шупика, м. Київ, Україна
Рубрики: Эндокринология
Разделы: Справочник специалиста
Версия для печати
Резюме
У першій частині лекції наведені сучасні дані про поширеність чоловічого гіпогонадизму, методи діагностики різних форм гіпогонадизму; описано клінічну характеристику найбільш поширених форм цієї патології.
В первой части лекции приведены современные данные о распространенности мужского гипогонадизма, методы диагностики различных форм гипогонадизма; описана клиническая характеристика наиболее распространенных форм данной патологии.
The first part of the review presents the current data on the prevalence of male hypogonadism, methods of diagnosing different forms of hypogonadism, describes the clinical manifestations of the most common forms of this disease.
Ключевые слова
гіпогонадизм; тестостерон
гипогонадизм; тестостерон
hypogonadism; testosterone
Чоловічий гіпогонадизм (ГГ) — загальна ендокринна проблема. Так, у США у 4–5 млн чоловіків діагностується ГГ. Близько 20 % чоловіків віком понад 60 років мають рівні тестостерону в крові нижчі від нижньої границі референсних значень, що може призводити до розвитку тестостеронової недостатності (ТН). Рецепти на продаж препаратів тесто–стерону в 1988 р. були виписані на 18 млн доларів; у 2011 р. витрати на них зросли до 1,6 млрд доларів із подальшим збільшенням. У 1992–2002 рр. частота виписування препаратів тестостерону збільшувалася на 25–30 % щорічно, а використання тесто–стерону в чоловіків віком понад 40 років зросло із 0,81 до 2,91 % за цей період. У той же час передбачають, що реальна частка ГГ в чоловіків залишається недіагностованою. Проблемними питаннями залишаються порогові рівні тестостерону в крові в чоловіків старших вікових груп для призначення тестостеронової терапії, а також її тривалість і співвідношення ефективності/безпеки.
За останню декаду вийшло декілька рекомендацій міжнародних та європейських асоціацій стосовно діагностики та лікування різних форм чоловічого ГГ (останні в 2016 р.), що свідчить про актуальність цієї проблеми.
Рекомендації Endocrine Society Clinical Practice визначають чоловічий ГГ як клінічний синдром, спричинений недостатньою продукцією фізіологічних рівнів тестостерону, зумовленою порушеннями на одному або декількох рівнях гіпоталамо-гіпофізарно-тестикулярної системи [1], причому андрогенова недостатність може несприятливо впливати на функції багатьох органів і систем та на якість життя [2].
Андрогени відіграють надзвичайно важливу роль у розвитку та підтриманні репродуктивної та сексуальної функцій у чоловіків; вони необхідні для розвитку чоловічих репродуктивних органів, процесу пубертації, ініціації та підтримки фертильності, композиції тіла, м’язів, мінералізації кісток, жирового метаболізму, когнітивних та поведінкових реакцій [3].
Загальний принцип програми формування статевого диференціювання полягає у формуванні плода за чоловічим типом і потребує активної дії генетичних (продуктів Y-хромосоми) і гормональних чинників (продуктів секреції тестикул ембріона). Основну роль в диференціюванні яєчок відіграє експресія численних генів, локалізованих на короткому плечі Y-хромосоми, включаючи секс–детермінуючий регіон Y-хромосоми. Яєчка плода продукують три гормони: тестостерон, інсуліноподібний пептид 3 (ІПП-3) і антимюллерів гормон (АМГ) [4]. Тестостерон метаболізується в більш потужний дигідротестостерон (ДГТ) та естрадіол [5]. Тестостерон і ДГТ необхідні для формування чоловічих зовнішніх статевих органів, передміхурової залози й сім’яних пухирців. АМГ необхідний для регресії мюллерових протоків, а інсуліноподібний пептид 3 разом з АМГ регулюють опущення яєчок. Інтратестикулярний тестостерон необхідний для підтримання сперматогенезу, причому його концентрація в 25–100 разів перевищує циркулюючий рівень гормону в крові. Дія тестостерону на гермінативні клітини опосередкована клітинами Сертолі через експресію андрогенового рецептора й вплив на мікрооточення у звивистих канальцях [6]. Продукція тестостерону контролюється лютропіном (ЛТ).
Функціонування гіпоталамо-гіпофізарно-статевої системи (ГГСС) кардинально відрізняється від подібних систем регуляції інших ендокринних органів. Унікальна особливість полягає у хвилеподібному характері її активності. У хлопчиків при народженні ГГСС функціонує на достатньо зрілому рівні, у препубертатний період спостерігається гальмування її активності, а в пубертатний період відбувається перебудова функціонування ГГСС, яка забезпечує вікові морфофункціональні перетворення в органах та системах організму. Таким чином, до пубертації рівні тестостерону є низькими, що попереджує вірилізацію. Головним компонентом статевого дозрівання є зростання активності статевих залоз, що призводить до підвищення секреції тестостерону. Підвищення секреції статевих стероїдів призводить до розвитку вторинних статевих ознак, зовнішніх і внутрішніх статевих органів, зміни пропорцій тіла й прискорення зросту, формування мінералізації кісткової тканини.
Тестостерон — найбільш важливий і фізіологічно активний андроген, який продукується яєчками. Він впливає практично на всі органи й системи організму (Swerdloff R., Wang Ch., 1989).
Фізіологічні ефекти тестостерону
Внутрішньоутробні:
— диференціація чоловічого статевого тракту,
— ріст статевого члена.
У період пубертації та в дорослого чоловіка:
— ріст волосся на лобку, в аксилярних западинах, на обличчі,
— розвиток зовнішніх статевих органів,
— ріст передміхурової залози та сім’яних пухирців,
— ріст тіла,
— сперматогенез (ініціація та підтримання),
— збільшення гортані та потовщення голосових зв’язок,
— розвиток м’язів,
— андроїдний розподіл підшкірно-жирової тканини,
— еритропоез,
— формування чоловічої психіки, підтримка лібідо та потенції.
Тестостерон здійснює свій вплив через андрогеновий рецептор, локалізований в цитоплазмі і ядрі клітини-мішені. Дефекти й мутації в гені андрогенового рецептора можуть призводити до порушення статевого розвитку, що може бути причиною розвитку тестикулярної фемінізації або слабо вираженої вірилізації, андрогенної резистентності та чоловічого безпліддя [7]. В екзоні першого гена є декілька ділянок із послідовностями, що повторюються, причому одна з них складається з 21 ± 2 повтори триплету CAG — цистеїн-аденозин-гуанозин. Встановлено, що зменшення числа повторів у чоловіків призводить до частішого розвитку гіпогонадизму, підвищеного ризику захворювань передміхурової залози, а також впливає на андрогенні фенотипові ефекти навіть у чоловіків із нормальними рівнями тестостерону [8].
Гіпогонадизм у чоловіків розвивається в результаті дисбалансу в ГГСС або рецепторних порушень в органах-мішенях. Залежно від рівня ураження системи розрізняють [3, 9]:
— первинний або гіпергонадотропний гіпогонадизм (яєчка),
— вторинний або гіпогонадотропний гіпогонадизм (гіпоталамус і гіпофіз),
— віковий гіпогонадизм (тестостеронова недостатність; гіпоталамус/гіпофіз і яєчка),
— андрогенорезистентність — органи-мішені андрогенів.
Отже, гіпогонадизм у чоловіків розвивається в результаті вроджених або набутих аномалій розвитку яєчок, що є причиною первинного гіпогонадизму, або порушень центральної нервової системи (ЦНС) та гіпоталамо-гіпофізарних структур, що є причиною вторинного гіпогонадизму.
Вроджені аномалії розвитку перерахованих вище ланок ГГСС у більшості випадків зумовлені хромосомними дефектами та генними мутаціями.
Набуті форми первинного гіпогонадизму розвиваються внаслідок інфекційних та автоімунних захворювань яєчок, травм, хірургічних втручань, променевої та хіміотерапії, прийому протипухлинних препаратів, наркотиків, токсичних речовин.
Набуті форми вторинного гіпогонадизму розвиваються в результаті інфекційних процесів, пухлин гіпоталамо-гіпофізарної ділянки та ЦНС, травм, хірургічних втручань із приводу пухлин гіпофіза, променевої терапії.
В основі первинного гіпогонадизму лежить ураження яєчок, що призводить до порушення їх здатності продукувати статеві гормони, зниження рівнів тестостерону в крові й, відповідно, до механізму зворотного зв’язку гіперстимуляції аденогіпофіза, що зумовлює підвищення рівнів гонадотропних гормонів.
В основі вторинного гіпогонадизму лежать пошкодження ЦНС і структур гіпоталамо-гіпофізарної системи, що призводять до зниження здатності гіпоталамуса секретувати гонадоліберин або гіпофізарні ЛТ і ФСГ, зумовлюючи зменшення продукції гормонів яєчками.
Первинне ураження гонадотропних клітин гіпофіза часто поєднується з недостатністю інших тропних гормонів: соматотропного, тиреотропного та адренокортикотропного.
Розвиток клінічних симптомів гіпогонадизму залежить від віку, в якому вперше розвинувся дефіцит андрогенів, та ступеня недостатності статевих залоз. Дефіцит тестостерону на ранніх стадіях ембріогенезу призводить до розвитку чоловічого псевдогермафродитизму, а на більш пізніх стадіях — до дистонії яєчок та мікропенісу [10]. Допубертатний гіпогонадизм характеризується недорозвитком статевих органів, відсутністю або слабкою вираженістю вторинних статевих ознак та формуванням євнухоїдного синдрому. Постпубертатний гіпогонадизм характеризується зниженням потенції та редукцією вторинних статевих ознак.
У табл. 1 наведені клінічні симптоми допубертатного й постпубертатного гіпогонадизму в підлітків [11].
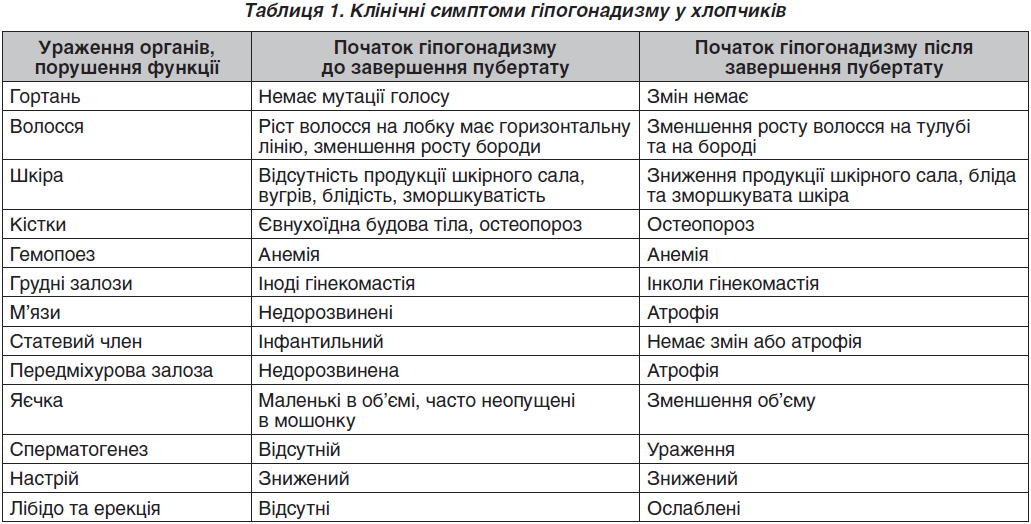
Вважають, що для препубертатного гіпогонадизму характерною є також затримка кісткового віку, центрипетальне ожиріння, об’єм яєчок
< 5 мл, довжина статевого члена < 5 см, а для пост–пубертатного — нормальні пропорції скелета, центрипетальне ожиріння, нормальна довжина статевого члена, об’єм яєчок < 15 мл [12].
У хлопчиків спостерігається різний ступінь затримки розвитку зовнішніх статевих органів уже в допубертатному періоді. Яєчка зменшені в розмірі, за консистенцією м’які або ущільнені. Часто спостерігається крипторхізм. Статевий член менший за розміром, характерним для вікової норми, у деяких випадках спостерігається мікропеніс. У пубертатному періоді не спостерігається збільшення об’єму яєчок (об’єм яєчок не перевищує 3–4 мл); не розвиваються або слабко розвиваються вторинні статеві ознаки: відсутні мутація голосу й оволосіння на лиці, ювенільне акне, ерекції та полюції. У хворих на гіпогонадизм уже в пубертатному віці починають формуватися євнухоїдні, гіноїдні або інфантильні пропорції тіла та остеопороз. Для первинного препубертатного гіпогонадизму характерними є євнухоїдно-гіноїдна будова тіла, збільшення розмірів кистей, стоп, нижньої частини лиця (субакромегалоїдність), помірний недорозвиток оволосіння обличчя, аксилярних западин і лобка, зменшення в розмірах статевого члена на тлі вираженої гіпоплазії яєчок та їх щільної консистенції.
У більшості підлітків, які страждають від гіпогонадизму, швидкість росту в препубертатний період нормальна або спостерігається незначне зниження в пубертатному періоді в результаті відсутності ростового стрибка. Здебільшого кінцевий зріст хворих із гіпогонадизмом не страждає і навіть перевищує генетично детермінований, оскільки зони росту довго лишаються відкритими, а спроможність до лінійного росту зберігається до 18–20 років та більше. Кістковий вік у таких пацієнтів спонтанно досягає межі 13–14-річних хлопчиків, і тільки після цього відзначається затримка кісткового дозрівання. У постпубертатний період така затримка може виявитися дуже суттєвою.
Однак у дітей із синдромами Шерешевського — Тернера, Нунан, із множинним дефіцитом тропних гормонів гіпофіза значна затримка зросту й кісткового віку спостерігається вже в допубертатному періоді, і без додаткового лікування хворі залишаються низькорослими [13].
У постпубертатному періоді зовнішні й внутрішні геніталії залишаються недорозвиненими; оволосіння бідне, характерним є його розподіл за жіночим типом; погано розвинені скелетні м’язи; розвивається еректильна дисфункція, олігозооспермія або азооспермія.
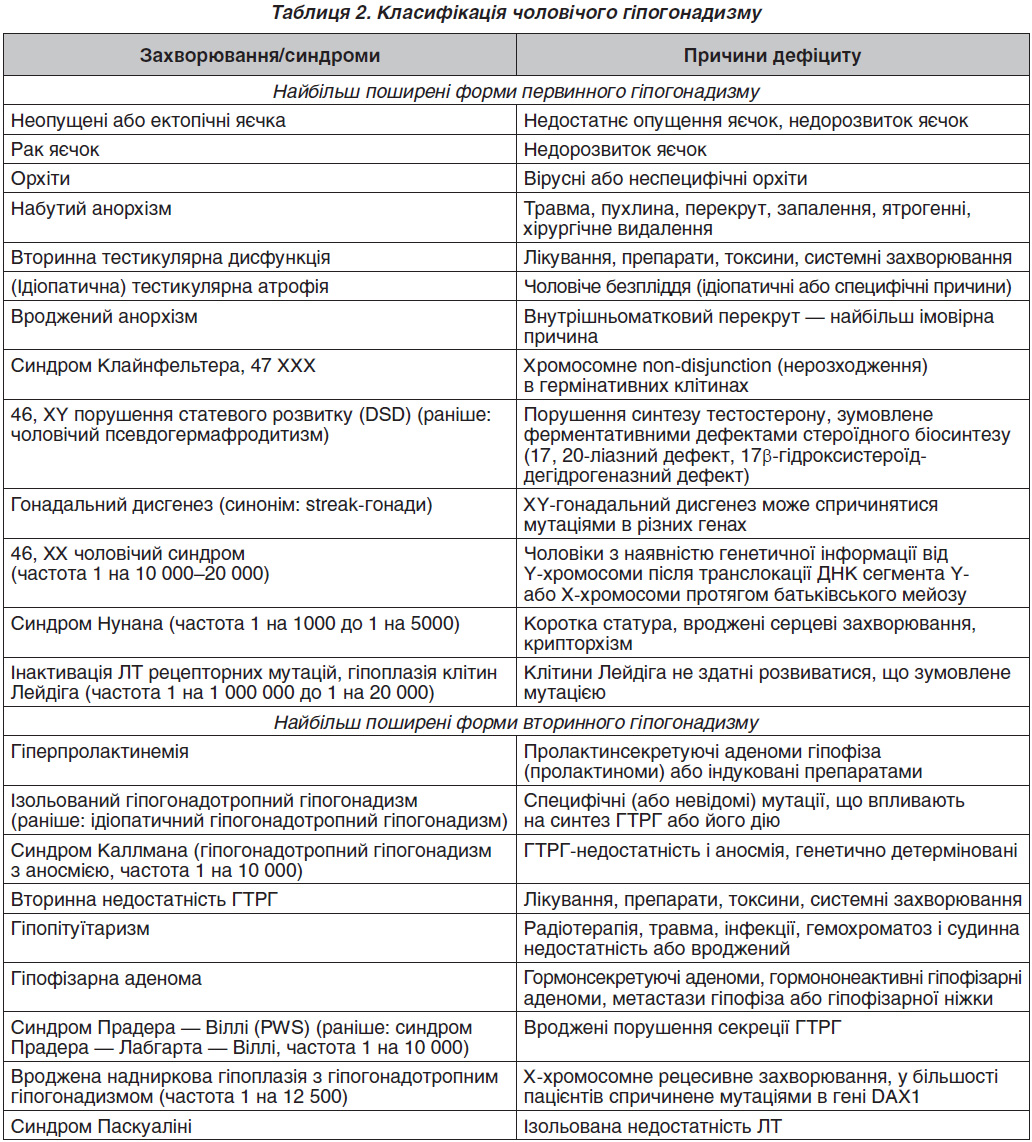
У рекомендаціях Європейської асоціації урологів запропонована така класифікація чоловічого гіпогонадизму (табл. 2) [3].
Первинна тестикулярна недостатність — найчастіша причина гіпогонадизму в чоловіків. Найбільш частою клінічною формою первинного гіпогонадизму є синдром Клайнфельтера, який спостерігається в 0,2 % популяції чоловіків. Захворювання зумовлене аномалією статевих хромосом, причому в 90 % випадків відзначається каріотип 47ХХХ, а також 48ХХYY, 48XXXY і мозаїчний тип 46XY/47XXY, 47ХХY/46ХХ, 47ХХY/45ХО. Наявність додаткової Х-хромосоми в чоловічому каріотипі може бути зумовлена нерозходженням Х-хромосоми в період першого чи другого мейотичного поділу або порушенням мітотичного розходження хромосом у період розвитку зиготи (мозаїчні варіанти) [14]. Частота народження хлопчиків із синдромом Клайнфельтера зростає зі збільшенням віку матері. Наявність додаткової Х-хромосоми в чоловічому каріотипі не впливає на диференціювання яєчок і формування статевих органів за чоловічим типом, але призводить до порушення гермінативних клітин і відсутності сперматогенезу. Останнім часом було встановлено наявність поодиноких сперматозоїдів у дорослих чоловіків із нормальним гаплоїдним хромосомним набором.
Синдром Клайнфельтера характеризується високим зростом пацієнтів, зменшенням розмірів яєчок, наявністю гінекомастії, безпліддям. У хлопчиків допубертатного віку відзначаються маленькі розміри яєчок та статевого члена, може виявлятися крипторхізм (частіше двобічний). У 50 % хлопчиків спостерігається помірна затримка розумового розвитку, яка супроводжується порушенням поведінки, тяжкістю контактів з однолітками. У пубертатному віці вторинне оволосіння виникає у звичайні строки, відзначається також збільшення статевого члена. Однак об’єм яєчок зростає незначно і зазвичай не перевищує 8 мл, причому яєчка мають щільну консистенцію. Фенотип у хворих чоловічий, характерні високорослість, непропорційно довгі ноги, євнухоїдна будова тіла, гінекомастія. Відставання кісткового віку від паспортного починається в 13–14 років. Зони росту залишаються відкритими до 18–20 років. Вторинне оволосіння скудне, за жіночим типом. У постпубертатному періоді яєчка залишаються маленькими (довга вісь менше ніж 2 см), щільними. При біопсії яєчка знаходять гіаліноз сім’яних канальців, зменшення числа або відсутність клітин Сертолі; сперматогенез відсутній.
За наявності мозаїцизму спостерігаються менш тяжкі порушення. У чверті хворих із мозаїцизмом яєчка мають нормальні розміри. У частини пацієнтів сперматогенез збережений.
Окрім симптомів порушення статевого розвитку, у хворих із синдромом Клайнфельтера може виявлятися низка вроджених аномалій кісткової тканини: клінодактилія, деформація грудини, вальгусні деформації ліктьових та колінних суглобів, гіпертелоризм, мікрогнатія, готичне піднебіння. Нерідко захворювання супроводжується вродженими вадами серцево-судинної системи. У дослідженнях останніх років у них встановлено підвищений ризик розвитку ЦД 2-го типу та несприятливі зміни в складі організму: накопичення жирової клітковини, зменшення м’язової маси, зниження чутливості до інсуліну та менший об’єм споживання кисню.
У чоловіків iз синдромом Клайнфельтера ризик раку молочної залози в 20 разів вищий, ніж у чоловіків із нормальним каріотипом.
Нерідко причиною первинного гіпогонадизму в чоловіків є пухлини яєчок, які діагностуються в молодих чоловіків після пубертації [15]. За останні 40 років частота раку яєчок у молодих чоловіків зросла вдвічі. Чинниками ризику є контра–латеральний рак гермінативних клітин, неопущені яєчка, гонадальний дисгенез, безпліддя, атрофія яєчок, сімейний рак гермінативних клітин. Класичним методом лікування пухлин яєчок є радикальна орхіектомія, а рівень вилікування за умов виявлення захворювання на ранніх стадіях становить понад 95 %. Після лікування в 22 % пацієнтів із пухлинами яєчок розвивається тестостеронова недостатність [16]. Cлід зазначити, що останнім часом виникла тенденція виконувати органозберігаючі операції в чоловіків із раком яєчок у певних випадках з метою уникнення розвитку неплідності та зниження продукції тестостерону.
Синдром Нунан — автосомно-рецесивне захворювання, зумовлене експресією патологічного гена, який локалізується на довгому плечі 12-ї хромосоми. Клінічна картина при синдромі Нунан характеризується низькорослістю, бочкоподібною грудною кліткою, укороченою шиєю з крилоподібними складками, лімфатичними набряками кистей та стоп, вальгусною деформацією суглобів, деформацією вушних раковин. У хлопчиків часто спостерігається крипторхізм, гіпоплазія яєчок, мікропеніс.
Типовою для синдрому Нунан є наявність у хворих кардіоваскулярних вад розвитку: стеноз легеневої артерії, який спостерігається у 80 % хворих, гіпертрофія міжшлуночкової перегородки. Відзначаються порушення сперматогенезу (оліго- або азооспермія).
ХХ-синдром у чоловіків, або синдром де ля Шапеля, трапляється в осіб із чоловічим фенотипом та каріотипом ХХ (1 на 9000). Припускають, що розвиток синдрому зумовлений транслокацією малої частини Y-хромосоми на Х-хромосому або на автосому. Можливо, інтактна Y-хромосома зберігається в одній із стовбурових клітин.
Клінічна картина характеризується гіпоплазованими яєчками в правильно сформованій мошонці, оволосіння розвивається за жіночим типом. Потенція в більшості випадків не порушена, але спостерігається гіпоспермія та азооспермія. Дуже рідко відзначається затримка розумового розвитку.
Вроджений анорхізм — відсутність яєчок у мошонці в нормальних за генотипом та фенотипом хлопчиків. Вважають, що різкий внутрішньоматковий перекрут є найбільш імовірною причиною захворювання. Клінічна картина: у пубертатному періоді у хлопчиків не розвиваються вторинні статеві ознаки, спостерігається євнухоїдний скелет, часто — ожиріння. Розміри мошонки, статевого члена, передміхурової залози не відрізняються від таких у інших дітей. Голос високий і тонкий, зменшена маса м’язів, євнухоїдні пропорції тіла, відсутня статева поведінка.
Набутий анорхізм розвивається в результаті травми, пухлини, перекруту, запалення, хірургічного видалення яєчок або буває ятрогенного походження. Клінічна картина залежить від віку пацієнта: у хлопчиків допубертатного періоду відсутнє статеве дозрівання й симптоми подібні до тих, які спостерігаються при вродженому анорхізмі, а в чоловіків у постпубертатному періоді відзначається еректильна дисфункція, безпліддя, поступове зниження вираженості вторинних статевих ознак.
Змішана дисгенезія гонад (змішаний гонадальний дисгенез, ЗГД) — одна з найбільш поширених форм несправжнього чоловічого гермафродитизму, яка становить 25–30 % від його усіх форм. Характеризується наявністю дисгенетичного яєчка, яке розташовується в черевній порожнині, паховому каналі або в рудиментарній мошонці. У більшості хворих виявляється мозаїчний каріотип 45ХО/46ХY. Клінічна картина: будова зовнішніх і внутрішніх статевих органів у хворих на змішаний гонадальний дисгенез залежить від клону клітин, який переважає у каріотипі, та від ступеня ураженості Y-хромосоми.
У пацієнтів із превалюванням клону ХY Y-хромосома суттєво не пошкоджена, тому яєчко функціонує у внутрішньоутробному періоді й спостерігається змішана будова зовнішніх статевих органів.
У пацієнтів із превалюванням клону ХО Y-хромосома суттєво пошкоджена, яєчко не функціонує у внутрішньоутробному періоді, тому характерною є більш жіноча будова зовнішніх статевих органів.
У всіх пацієнтів відзначається єдине яєчко, що має ознаки дисгенезії різного ступеня, спостерігається порушення його сперматогенної функції, але у частини хворих можливість яєчка продукувати тестостерон зберігається (функціонуюче яєчко), тому в більшості випадків спостерігається неповне статеве дозрівання.
У таких пацієнтів є великий ризик розвитку злоякісних пухлин із дисгенетичного яєчка, а поява симптомів андрогенізації може бути першою ознакою її розвитку.
Статевий хроматин у хворих негативний або показник дуже низький.
У пацієнтів із позитивним результатом функціонального тесту з хоріонічним гонадотропіном (ХГ) можливий спонтанний пубертат.
Однією з нечастих форм, яка відсутня в даній класифікації, є синдром дель Кастильо (Сертолі-клітинний синдром), що характеризується відсутністю зародкових клітин.
Основний симптом синдрому дель Кастильо — гіпоплазовані яєчка. Статевий член та мошонка розвинуті нормально, вторинні статеві ознаки також розвинуті достатньо, будова тіла нормальна. Потенція збережена, але хворі є неплідними.
Набуті форми гіпергонадотропного гіпогонадизму
Набуті форми гіпергонадотропного гіпогонадизму можуть розвиватися в результаті перенесених травм, хірургічних втручань (оперативне лікування крипторхізму, пахової грижі, перекруту яєчок), променевої терапії, хіміотерапії, інфекційних захворювань (епідемічний паротит з орхітом, туберкульоз), автоімунного процесу, що призводить до атрофії яєчок. Клінічна картина залежить від віку пацієнта — препубертатний чи постпубертатний.
Крипторхізм — захворювання, при якому одне або обидва яєчка не опущені в мошонку. Розрізняють одно- і двобічний, справжній і псевдокрипторхізм та ектопію яєчка.
При справжньому крипторхізмі яєчко знаходиться в будь-якому місці на шляху свого нормального опускання (у черевній порожнині, паховому каналі або перед паховим кільцем). При псевдокрипторхізмі яєчко може підніматися в паховий канал або в черевну порожнину. Цей стан діагностується вдвічі частіше, ніж справжній. При ектопії яєчко розташовується поза шляхом нормального опущення й зазвичай не пов’язане з грижовим мішком.
Частота крипторхізму серед доношених новонароджених хлопчиків становить 2–4 %, а в недоношених — 9–30 %. Після першого року життя поширеність крипторхізму становить від 0,4 до 2 %, а у підлітків — 0,3–3 %. Крипторхізм трапляється в 0,2–0,3 % дорослих чоловіків.
Виділяють три групи чинників патогенезу крипторхізму: механічні, гормональні, ендогенні.
Основним симптомом крипторхізму є відсутність яєчок у мошонці, недорозвиток і асиметрія мошонки. Іноді пацієнти відзначають біль у ділянці неопущеного яєчка. У багатьох хворих спостерігається поєднання крипторхізму з паховою грижею, диспластичними симптомами, ожирінням, недорозвитком статевого члена, порушеннями статевого розвитку, сперматогенезу. Поява вторинних статевих ознак запізнюється в половини підлітків із крипторхізмом. Крипторхізм, особливо черевний, є чинником підвищеного ризику виникнення раку яєчка, який розвивається в 3–16 % чоловіків, які в анамнезі мали крипторхізм.
Вроджені форми гіпогонадотропного гіпогонадизму
Для вторинного препубертатного гіпогонадизму характерна гіноїдно-євнухоїдна будова тіла: зменшення розмірів кистей, стоп, нижньої частини обличчя (субнаноїдність) із вираженою фемінізацією морфотипу, гіпотрофія статевого члена та наявність транзиторного фімозу. Інфантильний, або гіноїдний, морфотип спостерігається нечасто. У таких хлопчиків яєчка мають дряблу консистенцію, підтягнуті до пахових кілець.
Найбільш частою формою гіпогонадотропного гонадизму є синдром Каллмана.
Синдром Каллмана — вроджене захворювання, яке характеризується поєднанням гіпогонадотропного гіпогонадизму й аносмії або гіпоосмії. Це найчастіша причина гіпогонадотропного гіпогонадизму у хлопчиків, його частота перебуває у межах 1 : 5000–10 000 у популяції чоловіків. Синдром Каллмана становить собою третинну (гіпоталамічну) форму гіпогонадизму й зумовлений вродженим дефектом розвитку гіпоталамуса, що призводить до дефіциту ліберинів і зниження секреції гонадотропінів і тестостерону. Дефект формування нюхових нервів викликає аносмію або гіпоосмію. Розрізняють три типи синдрому Кал–лмана, зумовлені мутаціями гена KALIG-1 (KAL1), дефектами гена FGFR1 (KAL2) на короткому плечі восьмої хромосоми та генетичною гетерогенністю з автосомно-рецесивним, або Х-зчепленим рецесивним, або автосомно-домінантним типом спадковості. При всіх варіантах спостерігається порушення міграції гонадоліберинсекретуючих нейронів. Клінічна картина характеризується симптомами гіпогонадизму та євнухоїдизму, який нерідко супроводжується крипторхізмом, аносмією або гіпоосмією, соматичними дефектами розвитку («заяча губа»), розщепленням піднебіння («вовча паща»), високим піднебінням (готичне), шестипалістю, асиметрією язика, укороченням вуздечки язика, сенсорно-нервовими порушеннями слуху, спастичними параплегіями, мозочковою атаксією, горизонтальним ністагмом, порушенням кольорового зору, розумовою відсталістю, ураженням серцево-судинної системи, вадами розвитку сечостатевої системи. Клінічні прояви синдрому Каллмана можуть бути досить варіабельними. Хворі з синдромом Каллмана високорослі, мають євнухоїдні пропорції тіла. У пубертатному періоді відсутній ростовий стрибок, однак зони росту залишаються відкритими до 20 років і понад, тому лінійний зріст пацієнтів не страждає. Відставання кісткового віку від паспортного починається в 13–14 років.
Ізольований ідіопатичний гіпогонадотропний гіпогонадизм (ІІГГ). Захворювання спостерігається рідше, ніж синдром Каллмана. Припускають, що тотальний дефіцит продукції гонадотропінів у таких пацієнтів зумовлений ураженням гіпоталамуса. Мутації гена гонадоліберину ідентифіковані в 40 % пацієнтів. При ізольованому ІІГГ спостерігається дефіцит продукції ЛТ і ФСГ або одного з цих гормонів. Клінічна картина ІІГГ характеризується класичними ознаками препубертатного гіпогонадизму.
Синдром Паскуаліні (ізольований дефіцит лютропіну). Причиною розвитку захворювання є ізольований дефіцит лютропіну, що призводить до недостатньої секреції тестостерону яєчками. Клінічна картина захворювання залежить від вираженості дефіциту лютропіну: євнухоїдні пропорції тіла, недорозвиток статевого члена і яєчок, вторинних статевих ознак, порушення статевих функцій, високорослість. Відставання кісткового віку від паспортного починається в 13–14 років, але зони росту залишаються відкритими до 18–20 років. Фертильність у більшості пацієнтів із синдромом Паскуаліні порушена — зменшення об’єму сперми, кількості та рухливості сперматозоїдів. Захворювання з автосомно-домінантним типом успадкування. Частота синдрому невелика й становить 1 : 25 000 у популяції.
Клінічна картина синдрому Прадера — Віллі характеризується малою масою тіла в новонароджених дітей, вираженою м’язовою гіпотонією, маленькими розмірами кистей та стоп з укороченими пальцями, низькорослістю, низьким інтелектом, крипторхізмом, мікропенісом. Форма обличчя характеризується звуженим фронтальним діаметром, очі близько розташовані, міміка лиця слабо виражена. У процесі росту дитини розвивається ожиріння, зумовлене поліфагією. Клінічні симптоми гіпогонадотропного гіпогонадизму в дітей обох статей проявляються в пубертатному віці.
Гіпогонадотропний гіпогонадизм розвивається також у пацієнтів із синдромом Лоуренса — Муна — Барде — Бідля, який відсутній в даній класифікації. Це захворювання за автосомно-рецесивним типом успадкування. Клінічна картина синдрому Лоуренса — Муна — Барде — Бідля характеризується ожирінням, тотальним або частковим гіпогонадотропним гіпогонадизмом, пігментним ретинітом, спастичною параплегією, у 70–75 % хворих — полі- та синдактилія, вроджені аномалії нирок. Клінічні симптоми гіпогонадизму проявляються в пубертатному віці досить рано у вигляді різкого недорозвитку зовнішніх статевих органів.
Вроджена гіпоплазія надниркових залоз і гіпогонадотропний гіпогонадизм — ізольована форма гіпогонадотропного гіпогонадизму в чоловіків із вродженою гіпоплазією надниркових залоз. Вроджене захворювання, зумовлене мутацією гена DAX1, який відіграє ключову роль у розвитку надниркових залоз і ГГСС. Це сімейне захворювання, Х-зчеплений тип успадкування.
Клінічна симптоматика характеризується тяжкою глюкокортикоїдною недостатністю, синдромом втрати солі та симптомами гіпогонадотропного гіпогонадизму, які проявляються в пубертатному віці.
Набуті форми гіпогонадотропного гіпогонадизму
Основною причиною набутих форм гіпогонадотропного гіпогонадизму є пухлини, травми, інфекційно-запальні процеси гіпоталамо-гіпофізарної системи. Пухлини можуть призводити до зниження не тільки секреції гонадотропних гормонів, але й інших тропних функцій, в першу чергу соматотропної, що супроводжується різким зниженням швидкості росту пацієнтів. Ураження задніх відділів гіпофіза викликає розвиток нецукрового діабету. Досить часто пухлини призводять до порушень зору, зумовлених здавленням або руйнуванням оптичної хіазми, — звуження полів зору, атрофії дисків зорового нерва.
Гіперпролактинемічний гіпогонадизм зумовлений пролактинсекретуючими пухлинами гіпофіза — макро- і мікропролактиномами, препаратами з дофамінантагоністичними ефектами (метоклопрамід, іміпрамін, фенотіазин), а також може спостерігатися в пацієнтів із хронічною нирковою недостатністю й гіпотиреозом. Патогенез гіпогонадизму в пацієнтів із гіперпролактинемічним синдромом зумовлений пригніченням імпульсної секреції ЛТ-РГ, надлишковою секрецією пролактину, що викликає атрофію клітин Лейдіга та розвиток гіпогонадизму. Клінічна картина характеризується симптомами гіпогонадизму, зниженням лібідо, еректильною дисфункцією, гінекомастією, часто безпліддям, а також порушенням зору й неврологічною симптоматикою. Гіперпролактинемія, яка виникає в препубертатному або пубертатному періодах, може призводити до затримки статевого розвитку або гіпогонадизму. Клінічна вираженість андрогенної недостатності в чоловіків із відносно невеликим зниженням рівнів тестостерону в крові може бути зумовлена конверсією тестостерону в дигідротестостерон у периферичних тканинах.
Досить часто причиною набутого гіпогонадотропного гіпогонадизму в дітей є краніофарингеома — вроджена пухлина, яка розвивається в кишені Ратке, локалізується в ділянці турецького сідла, характеризується ендоселярним ростом, що призводить до випадіння функцій гіпофіза та розвитку гіпогонадизму, гіпотиреозу, нерідко з порушенням терморегуляції, нецукровим діабетом, порушенням зору, аносмією.
Променева терапія краніальної ділянки або загальна променева терапія при злоякісних пухлинах може призводити до розвитку пангіпопітуїтаризму, який також проявляється симптомами гіпогонадотропного гіпогонадизму.
До захворювань, зумовлених дефектами андрогенових рецепторів в органах-мішенях з повною, частковою або мінімальною андрогенорезистентністю (АР), відносяться синдром Рейфенштейна, синдром дефіциту 5-альфа-редуктази й хвороба Кеннеді (бульбоспінальна м’язова атрофія). Мутації андрогенових рецепторів знайдені в більшості пацієнтів із повною формою АР, але в 28–73 % — часткова АР [17].
Синдром дефіциту 5-альфа-редуктази є однією з форм несправжнього чоловічого гермафродитизму, каріотип 46ХY.
Фермент 5-альфа-редуктаза каталізує перетворення тестостерону в дегідротестостерон, який відіграє основну роль у процесах диференціювання зовнішніх статевих органів у чоловіків. Дефіцит 5-альфа-редуктази виникає в результаті мутацій гена SRD5A2, локалізованого на короткому плечі другої хромосоми з автосомно-рецесивним характером успадкування.
Фенотип у новонароджених наближається до жіночого. Урогенітальний синус містить короткий вагінальний відросток, який сліпо закінчується. Статевий член нагадує гіпертрофований клітор, кавернозні тіла нерозвинені. Внутрішні статеві органи сформовані за чоловічим типом: яєчка розвинені, розташовані в пахових каналах або скротолабіальних складках, виявляються сім’явиносні протоки, сім’яні пухирці, придаток сім’яника. Передміхурова залоза недорозвинена. У пубертатному віці підвищується концентрація тестостерону, що забезпечує виражену маскулінізацію зовнішніх геніталій та розвиток вторинних статевих ознак за чоловічим типом. Лабораторно визначаються нормальні рівні тестостерону, ЛТ, ФСГ у крові, низький рівень ДГТ. Співвідношення тестостерону і ДГТ суттєво підвищене (за рахунок зниження рівня ДГТ) і збільшується на тлі функціонального тесту з ХГ (за рахунок підвищення рівня тестостерону).
Синдром Рейфенштейна, або синдром неповної резистентності до андрогенів, зумовлений порушенням чутливості тканин до андрогенів та є генетично детермінованим захворюванням. Мутація гена, який відповідає за синтез рецептора до андрогенів, призводить до порушення взаємодії андрогенів із рецепторами та до резистентності. Описано декілька сотень мутацій гена, тому спектр клінічної симптоматики при синдромі досить широкий. Відсутня кореляція між фенотипом та генотипом у хворих із синдромом неповної резистентності до андрогенів. Клінічна картина характеризується симптомами гіпогонадизму: оволосіння на обличчі й у пахових ділянках слабо виражене, на лобку — за жіночим типом, яєчка дещо гіпоплазовані, мошонка розвинена нормально, статевий член недорозвинений, викривлений донизу (коротка вуздечка), у хворих нормостенічна будова тіла, часто спостерігається гіпоспадія, гінекомастія, крипторхізм, атрофія сім’яних канальців, безпліддя, хоча відзначається багато симптомів, подібних до синдрому Клайнфельтера. Генотип нормальний (46XY). Концентрація тестостерону, естрадіолу, ЛТ, ФСГ в крові підвищена. При гістологічному дослідженні біоптату яєчок знаходять гіалінізацію сім’яних канальців, достатню кількість клітин Лейдіга.
При повній нечутливості до андрогенів (синдром Моріса, або синдром тестикулярної фемінізації) спостерігається не чоловічий генотип (46XY), а жіночий.
Тестостеронова недостатність (віковий/пізній гіпогонадизм (late-onset hypogonadism)). Циркулюючі рівні тестостерону в крові знижуються прогресивно з віком, починаючи з другої і третьої декади життя, через порушення на всіх рівнях ГГСС, але траєкторія падіння залежна від ІМТ, прибавки маси тіла, коморбідних станів і генетичних чинників [18]. Клінічна картина ТН визначається як поєднання низьких рівнів тестостерону й клінічно суттєвих симптомів та є неспецифічною. Тому немає дефінітивного (чіткого, ясного) набору симптомів або профілю [19]. У чоловіків із низьким рівнем тестостерону в крові часто симптоми можуть розглядатися як асоціація між синдромом тестостеронового дефіциту й асоційованими коморбідними станами (цукровий діабет 2-го типу, серцево-судинні захворювання). У частини чоловіків середнього віку (~50–65 років) і старших чоловіків (> 65 років) залежне від віку падіння рівнів тестостерону асоціюється з кластером симптомів і ознак, подібних до тих, які спостерігаються в чоловіків з класичною андрогеновою недостатністю [19]. Залежні від віку референтні коливання рівнів тестостерону досі не визначені, і критерії та кінцеві рівні для діагностики ТН залишаються частково суперечливими. Поширеність біохімічного гіпогонадизму, за даними різних дослідників, становить від 23,3 % у чоловіків 40–70 років, за даними EMAS [18], та є залежною від віку.
На Міжнародній експертній консенсусній конференції у 2015 р. дійшли висновку, що ТН у чоловіків є серйозним медичним станом, асоційованим із несприятливим профілем серцево-судинного ризику, негативно впливає на чоловічу фізичну, психологічну та статеву функції, сприяє розвитку абдомінального ожиріння, цукрового діабету 2-го типу, синдрому втомлюваності, зменшенню кісткової тканини та маси й сили м’язів, анемії, депресії, підвищенню частоти еректильної дисфункції, зниженню якості життя [20].
Найбільш індикативні симптоми ТН у літніх чоловіків:
— статеві (зниження лібідо, ерекцій адекватних і спонтанних, статевої активності, статевих фантазій),
— фізичні (підвищений ризик обмеження рухливості, ламкості кісток і падіння, зниження маси й сили м’язів, підвищення відкладання жирових мас),
— психологічні (депресія, роздратованість, сонливість, порушення когнітивної функції) [21].
Конфлікт інтересів. Автори заявляють про відсутність конфлікту інтересів при підготовці даної статті.
Список литературы
1. Bhasin S., CunninghamG.R., Hayes F.J. et al. Testosterone therapy in men with androgen deficiency syndromes: an Endocrine Society Clinical Practice guideline // J. Clin. Endocrinol. Metabol. — 2010. — Vol. 95. — P. 2536-2539.
2. Nieschlag E. et al. Andrology: male reproductive health and dysfunction. 3rd ed. — Heidelberg, 2010.
3. Dohle G.R., Arver S., Bettocchi C. et al. EAU Guidelines on Male Hypogonadism. EAU, 2016. — 26 р.
4. Brinkman A.O. et al. Molecular mechanisms of androgen action — a historical perspective // Methods Mol. Biol. — 2011. — Vol. 776. — P. 3.
5. de Ronde W. et al. Aromatase inhibitors in men: effects and therapeutic options // Reprod. Biol. Endocrinol. — 2011. — Vol. 9. — P. 93.
6. McLachlan R.I. et al. Hormonal regulation of spermatogenesis in primates and man: insights for development of the male hormonal contraceptive // J. Androl. — 2002. — Vol. 23. —
P. 149.
7. Zitzmann M. Mechanisms of disease: pharmacogenetics of testosterone therapy in hypogonadal men // Nat. Clin. Pract. Urol. — 2007. — Vol. 4. — P. 161.
8. Canle D. et al. Androgen receptor polymorphism (CAG repeats) and androgenicity // Clin. Endocrinol. (Oxf). — 2005. — Vol. 63. — P. 356.
9. Nieschlag E. et al. Testosterone: action, deficiency, substitution. 2004. — Cambridge: University Press, 2004.
10. Greenspoon F.S., Strewler G.J. Basic and Clinical Endocrinology. 5th ed. — 1997. — P. 404-430.
11. Zitzmann M., Faber S, Nieschlag Е. Association of specific symptoms and metabolic risks with serum testosterone in older men // J. Clin. Endocrinol. Metab. — 2006. — Vol. 11. — P. 4335-4343.
12. Древаль А.В., Редькин Ю.А. Возрастной гипогонадизм: основные принципы диагностики и лечения // Российский медицинский журнал. — 2016. — 1. — 29-31.
13. Лучицький Є.В., Лучицький В.Є., Пилипенко В.М. Порушення статевого дозрівання. Клінічна ендокринологія дитячого та підліткового віку; за ред. М.Д. Тронька
і О.В. Большової. — К.: Здоров’я України, 2016. — С. 497-575.
14. Tuttelmann F. et al. Novel genetic aspects of Klinefelter’s syndrome // Mol. Hum. Reprod. — 2010. — Vol. 16. — P. 386.
15. Puhse G. et al. Testosterone deficiency in testicular germ-cell cancer patients is not influenced by oncological treatment // Int. J. Androl. — 2011. — Vol. 34. — P. 351.
16. Nord C. et al. Gonadal hormones in long-term survivors 10 years after treatment for unilateral testicular cancer //
Eur. Urol. — 2003. — Vol. 44. — P. 322.
17. Jaaskelainen J. Molecular biology of androgen insensitivity // Mol. Cel. Endocrinol. — 2011. — Vol. 352. — P. 4-12.
18. Tajar A., Huhtaniemi I.T., O’Neill T.W. et al. EMAS Group. Characteristics of androgen deficiency in late-onset hypogonadism: results from the European Male Aging Study (EMAS) // J. Clin. Endocrinol. Metabol. — 2012. — Vol. 97. — P. 1508-1516.
19. Lawrence K.L., Stewart F., Larson B.M. Approaches to male hypogonadism in primary care // The Nurse Pract. — 2017. — Vol. 42. — P. 32-37.
20. Wu F.C. et al. Identification of late-onset hypogonadism in middle-aged and elderly men // N. Engl. J. Med. — 2010. —
Vol. 363. — P. 123-135.
21. Morgentaler A., Zitzman M., Traish A.M. et al. Fundamental Concepts Regarding Testosterone Deficiency and Treatment: International Expert Consensus Resolutions // Mayo Clin. Proc. — 2016. — P. 1-16.