Вступ
Фіброз печінки — це локальне або дифузне збільшення кількості сполучної тканини, позаклітинного матриксу (колагенової волокнистої тканини в перисинусоїдному просторі), і розвиток фіброзу — це основний шлях прогресування хронічних дифузних захворювань печінки [2, 18, 47]. На ранніх стадіях фіброзу немає клінічних проявів, і лише при гістологічному дослідженні біоптату виявляється надмірне накопичення сполучної тканини. Надалі фіброз призводить до утворення вузлів регенератів, судинних анастомозів — формування цирозу печінки [58, 59].
Найбільш частими причинами розвитку цирозу печінки визнаються хронічна інтоксикація алкоголем (за різними даними, від 40–50 до 70–80 %) і вірусні гепатити В, С і D (30–40 %) [27, 30, 32, 36]. Найважливіші етапи виникнення алкогольного цирозу печінки — це гострий алкогольний гепатит і жирова дистрофія печінки з фіброзом і мезенхімальною реакцією. Особливо тяжким перебігом відзначаються алкогольно-вірусні цирози печінки з швидкопрогресуючою динамікою захворювання. Вони ж найбільш часто трансформуються в гепатоцелюлярну карциному [8, 33, 40, 49]. Значно рідше в розвитку цирозу печінки відіграють роль хвороби жовчних шляхів (внутрішньо- і позапечінкових), застійна недостатність серця, різні хімічні й лікарські інтоксикації.
Формування цирозу печінки відбувається протягом багатьох місяців або років. За цей час змінюється генний апарат гепатоцитів і формуються покоління патологічно змінених клітин. Цей процес у печінці можна охарактеризувати як імунозапальний. Найважливіший фактор у генезі алкогольного цирозу печінки — пошкодження (некроз) гепатоцитів, обумовлений прямою токсичною дією алкоголю, а також автоімунними процесами. Сенсибілізація імуноцитів до власних тканин організму — важливий фактор патогенезу і при цирозі, що розвивається у хворих на вірусні гепатити В, С і D. Основною мішенню автоімунної реакції є печінковий ліпопротеїд. Домінуючий фактор патогенезу застійного цирозу печінки — некроз гепатоцитів, пов’язаний із гіпоксією й венозним застоєм.
Подальшим етапом розвитку патологічного процесу є портальна гіпертензія — підвищення тиску в системі портальної вени, обумовлене обструкцією внутрішньо- або позапечінкових портальних судин. Портальна гіпертензія, у свою чергу, призводить до виникнення портокавального шунтування крові, спленомегалії й асциту [4, 9, 16].
Процеси фіброзу в печінці вивчалися протягом багатьох років, але тільки після з’ясування ролі зірчастих клітин у процесах фіброгенезу були відкриті нові можливості антифібротичної терапії [15, 19–21]. Синусоїдальні клітини — ендотеліальні, клітини Купфера, зірчасті клітини разом з розташованою в просвіті синусоїдів ділянкою гепатоцитів утворюють функціональну одиницю. Крім клітин, у ділянці синусоїдов розташовується позаклітинний матрикс (ПКМ), який видно тільки при захворюваннях печінки. Усі клітини, що утворюють синусоїд, можуть брати участь в утворенні ПКМ. У нормі існує рівновага між факторами фіброгенезу й антифібротичними факторами. Основну роль у фіброгенезі відіграють зірчасті клітини, що виробляють профібротичні й анти–фібротичні фактори [14, 46, 52, 53]. До антифібротичних факторів відносять матриксні металопротеїнази (ММП), що беруть участь у руйнуванні білків ПКМ (колагенази, желатинази, стромолізини) і синтезуються переважно макрофагами [3, 5, 6, 17, 39]. Активність ММП пригнічується тканинними інгібіторами матрикс–них металопротеаз (ТІММП), що також продукуються зірчастими клітинами.
При пошкодженні печінки виділяються біологічно активні речовини, що активують макрофаги й ендотелій синусоїдів, які виділяють інтерлейкін-1, фактор некрозу пухлини альфа (ФНП-α), оксид азоту, ендо–телін, діючи на зірчасті клітини. Зір–часті клітини при активації виробляють тромбо–цитактивуючий фактор (Platelet-derived growth factor, PDGF) і трансформуючий фактор росту (Transforming growth factor beta 1, TGF-β1). Під дією TGF-β1 зірчасті клітини починають активувати самі себе, мігрують у ділянки запалення. Відбувається зміна фенотипу зірчастих клітин: вони трансформуються в міофібробласти, що продовжують виробляти TGF-β1 і починають продукувати ПКМ. Порушення рівноваги між фібротичними й антифібротичними факторами веде до збільшення в 3–10 разів компонентів ПКМ, зміни його складу (переважання колагену I і III типу). Перерозподіл матриксу в простір Діссе, його розширення, капіляризація синусоїдів супроводжується порушенням обміну між гепатоцитами й кров’ю, шунтуванням крові через розвиток несправжніх часточок і розвитком цирозу печінки. У разі припинення дії медіаторів запалення зірчасті клітини знову починають продукувати антифібротичні речовини, і відбувається зменшення компонентів ПКМ у просторі Діссе. До антифібротичного каскаду підключаються макрофаги, які синтезують металопротеїнази, руйнують колагенові мікрофібрили й утилізують їх залишкові фрагменти [45, 51, 54, 55]. Отже, фіброз на ранніх стадіях розвитку — процес оборотний.
Патогенез фіброзу печінки при хронічних вірусних гепатитах пов’язаний з індукцією інфікованими гепатоцитами активності запальних клітин, що веде до стимуляції зірчастих клітин. При алкогольній хворобі печінки ацетальдегід і вільні радикали кисню також активують зірчасті клітини [34, 37]. Крім цього, етанол сприяє зростанню грамнегативної мікрофлори в кишечнику, підвищенню рівня ліпополісахаридів у портальній крові й активації клітин Купфера, які продукують ФНП-α, що діє на зірчасті клітини [48]. Патогенез фіброзу печінки при неалкогольній жировій хворобі печінки пов’язаний із гіперглікемією й інсулінорезистентністю, що ведуть до підвищення рівня вільних жирних кислот і розвитку стеатозу печінки, а дія вільних радикалів й прозапальних цитокінів призводить до некрозу гепатоцитів, виникнення вогнища запалення, у якому будуть експресуватися прозапальні цитокіни, що спричиняють прогресування фіброзу печінки. При первинному біліарному цирозі холангіоцити секретують фіброгенні медіатори, що активують зірчасті клітини, які запускають фіброгенез [23, 25, 26, 31].
Тривалий час фіброз печінки вважався необоротним патологічним станом. Однак ще 60 років тому були описані випадки зворотного розвитку фіброзу після ефективної терапії гемохроматозу й хвороби Вільсона, а в подальшому неодноразово публікувалися дані про зворотний розвиток фіброзу при автоімунному гепатиті в результаті імуносупресивної терапії, при вторинному біліарному цирозі печінки — після хірургічної декомпресії жовчовивідних шляхів, при неалкогольному стеатогепатиті — при зменшенні маси тіла, при алкогольному гепатиті — при абстиненції.
Оборотність фіброзу спостерігалася при тривалому утриманні від прийому алкоголю: через 4–6 тижнів було виявлено зменшення вмісту колагену IV типу, ламініну й гіалуронової кислоти в стінках синусоїдів при біопсії. Були відзначені також зміни, що відображають функцію зірчастих клітин: підвищення рівня ММП-2 і зниження рівня її інгібітору ТІММП-2. Через певні часові інтервали спостерігалося зменшення кількості міофібрил актину в стінках синусоїдів, що свідчить про падіння активності зірчастих клітин і переключення їх із синтезу екстрацелюлярного матриксу на його деградацію.
У той же час тільки з упровадженням у клінічну практику противірусної терапії концепція фіброзу печінки як динамічного процесу з можливістю як прогресування, так і регресу була доведена науково [12, 13, 22, 35, 41]. Аналіз позитивних клінічних результатів сформував чітке розуміння того, що фіброз печінки може регресувати. Ефективна антифібротична терапія може суттєво змінити принципи ведення пацієнтів із хворобами печінки й забезпечити сприятливий прогноз навіть за умов уже розвиненого цирозу печінки [10, 44, 56].
Довгий час золотим стандартом діаг–ностики фіброзу печінки було проведення біопсії з гістологічним дослідженням. Однак у зв’язку з інвазивністю, досить великою похибкою гістологічного дослідження, пов’язаною з «помилками попадання» голки при пункційній біопсії печінки, відмінністю в інтерпретації результатів для ранньої діагностики патологічних процесів у даний час велику увагу приділяють неінвазивним методам діагностики фіброзу. До них відносять біопрогностичні лабораторні тести; еластометрію печінки й магнітно-резонансну еластографію; ультразвукове дослідження (УЗД), комп’ютерну томографію, магнітно-резонансну томографію печінки, ультра–звукову допплерографію судин печінки й селезінки з розрахунком індексів фіброзу й портальної гіпертензії [7].
Антифібротична терапія нерозривно пов’язана з етіологічним і патогенетичним лікуванням хронічних гепатитів. У більшості випадків препарати для усунення етіологічних факторів гепатитів демонструють також і антифібротичну ефективність. Виявлено антифібротичну дію противірусних препаратів, пентоксифіліну, фосфатидилхоліну, глюкокортикостероїдів, донаторів оксиду азоту, вітаміну Е, антагоністів ендотелінових рецепторів, антагоністів ангіотензинових рецепторів, інгібіторів ангіотензинперетворюючого ферменту, силімарину.
Відбувається дослідження ефективності таких перспективних лікувальних методик із цілеспрямованою антифібротичною дією:
► елімінація пошкоджуючого агента (інтерлейкін-10, інгібітори ФНП — протизапальний ефект; антиоксиданти — пригнічення фібротичних процесів у відповідь на оксидативний стрес);
► гальмування профібротичної активності зірчастих клітин (інтерферони, фактор росту гепатоцитів, агоністи PPAR-γ);
► підтримання активної антифібротичної активності зірчастих клітин (антагоністи TGF-β1, що зменшують синтез матриксу й підсилюють його розпад; антагоністи PDGF, оксид азоту);
► вплив на секрецію колагену зірчастими клітинами печінки (інгібітори полігідроксилаз, інтерферон гамма — зменшують фіброз; антагоністи ендотелінових рецепторів — зменшують фіброз і портальну гіпертензію);
► дія на апоптоз зірчастих клітин (гілотоксин і фактор зростання нейронів стимулюють апоптоз);
► посилення розпаду колагенового матриксу (металопротеїнази, антагоністи тканинного інгібітору ММП; антагоністи TGF-β1 — знижують активність TІММП і підвищують активність ММП) [11, 38].
Перспективним є використання з антифібротичною метою і для регенерації тканин печінки стовбурових клітин, а також регуляторних пептидів натурального (рослинного й тваринного) походження, які за молекулярною структурою схожі на дефензини [24, 28, 43, 57]. Активація дефензинів та інших регуляторних пептидів у печінці прогнозовано приводить до підвищення ефективності етіотропної противірусної терапії гепатиту, а також сприяє очищенню органа від інфікованих клітин унаслідок активації регуляторних макрофагів. Регуляторні макрофаги, у свою чергу, продукують протизапальні інтерлейкіни, які налаштовують антифібротичну активність імунокомпетентних клітин і опосередковано підтримують регенераторні процеси в печінці [1, 29, 42, 50].
☼ Мета дослідження: вивчити антифібротичні властивості препарату Біірін і його вплив на результати лікування цирозу печінки інфекційної й токсичної етіології.
Матеріали та методи
На базі клініки печінкової патології «Регенерація» в період з 2015 по 2018 р. накопичений досвід застосування препарату Біірін у 24 пацієнтів (24 чоловіки віком від 44 до 65 років) із фіброзом і цирозом печінки. Фібротичні зміни в печінці розвивалися на тлі хронічного запалення внаслідок пошкодження паренхіми органа вірусами або токсинами. Відповідно, всі хворі були розподілені на дві групи: А і В. До групи А увійшли пацієнти з вірусним гепатитом і постінфекційним фіброзом печінки, у групу В — пацієнти з алкогольним гепатитом. Для підтвердження антифібротичних властивостей Бііріну були набрані контрольні групи пацієнтів — С (n = 30) і D (n = 30).
Антифібротичні властивості Бііріну вивчаються протягом останніх 10 років. Рослинні пептиди й наночастинки металів, що входять до складу Бііріну, потрапляючи в печінку, активують антиінфекційні білки вродженого імунітету, зокрема альфа- й бета-дефензини. Дефензини асистують лімфоцитам у виявленні інфікованих клітин і ліцензують макрофаги для очищення печінкової тканини від інфектів. Накопичення Бііріну в печінці приводить до синтезу протизапальних цитокінів і дестабілізації TGF-β1. На тлі зменшення запалення відбувається блокування патологічного фіброзного каскаду. Активуються регуляторні макрофаги й починається синтез матриксних металопротеїназ. Регуляторні макрофаги ініціюють апоптоз зірчастих клітин і міофібробластів. Знижується інтенсивність, а потім припиняється синтез колагенових мікрофібрил. Рештки фрагментів колагенових волокон під–даються фагоцитозу. Коли створено необхідні умови для відновлення повноцінної й здорової печінкової тканини, синтез Т-лімфоцитами інтерлейкіну-17 запускає механізми диференціювання гепатоцитів зі стовбурових клітин-попередників. Крім того, за рахунок модуляції синтезу регуляторних пептидів триває процес оновлення мембранних структур, нормалізується метаболізм та енергетичний обмін у клітинах. Оновлені гепатоцити більш стійкі щодо дії пошкоджуючих факторів і відзначаються підвищеною пластичністю.
Результати та обговорення
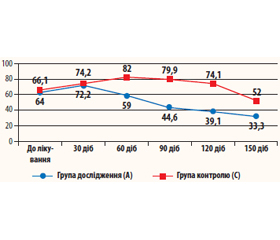
Клінічні спостереження показують, що у груп хворих із вірусним гепатитом (табл. 1, рис. 1) відбулося зростання рівня TGF-β1 на початковому етапі з подальшим його зниженням (нормалізацією). Проте у хворих, які приймали Біірін (група А), рівень TGF-β1 зростав тільки до 2-го візиту (з 64 до 72,2 нг/мл), а в подальшому поступово знижувався до 33,3 ± 6,0 нг/мл. Дана динаміка є цілком обґрунтованою, вона обумовлена антифібротичними механізмами, що активуються при прийомі препарату. У контрольній групі хворих із вірусним гепатитом (група С) латентний період був значно довшим, із поступовим підвищенням рівнів TGF-β1 до 60-ї доби лікування (з 66,1 до 82,0 нг/мл) і подальшим зниженням після 90-ї доби лікування.
Динаміка рівнів MMП-1 у групах А та С є порівнянною до 60-ї доби лікування з подальшим більш інтенсивним приростом у групі хворих, які приймали Біірін (А), — до 14,1 ± 2,5 нг/мл і суттєво меншою динамікою в контрольній групі С.
Статистично значимої різниці між групами А та С за рівнями аланінамінотрансферази (АлАТ) та аспартатамінотрансферази (АсАТ) не виявлено протягом усіх періодів спостереження. Проте нормалізація вказаних показників спостерігається практично з 30-ї доби лікування з дещо більш вираженою нормалізацією (наближення до нормальних рівнів) показників у групі А (Біірін). Гамма-глутаматтрансфераза (ГГТ) є більш чутливим показником порушень процесів у клітинах печінки, ніж АлАТ і АсАТ. За рівнями ГГТ статистично значима різниця (p < 0,05) із більш суттєвим покращенням показників у групі А виявляється із 120-го дня лікування. За динамікою загального білірубіну не виявлено суттєвої різниці між групами.
Аналіз у групах хворих з алкогольним гепатитом і наявністю фіброзу (група В з прийомом Бііріну й контрольна група D) виявив схожу динаміку досліджуваних клінічних параметрів (табл. 2, рис. 3, 4).
У групі В (Біірін) протягом курсової терапії рівень TGF-β1 знижувався з 66,0 ± 10,2 нг/мл до 34,1 ± 3,0 нг/мл. У той же час у контрольній групі D динаміка була значно меншою — з 69,2 ± 13,1 нг/мл до 51,3 ± 2,2 нг/мл. Статистично значима різниця (p < 0,05) між групами за рівнями TGF-β1 виявлялась уже після 1 місяця лікування.
Рівні MMP1 у групах B і D є порівнянними до 60-ї доби лікування з подальшим більш вираженим приростом у групі хворих, які приймали Біірін (В), — до 14,4 ± 2,3 нг/мл і суттєво меншою (p < 0,05) динамікою в контрольній групі (D).
Нормалізація рівнів АлАТ і АсАТ спостерігається практично з 30-го дня спостереження, проте не виявлено суттєвої різниці між групами за кожен період спостереження (p > 0,05).
Динаміка рівня загального білірубіну суттєво не відрізняється між групами.
Наявність статистично значимої різниці між групами дослідження як в окремі періоди спостереження, так і за динамікою клінічних показників обумовлює доцільність проведення поглибленого аналізу з порівнянням досягнутих клінічних ефектів лікування в групах і їх оцінкою.
Показник стандартизованої величини клінічного ефекту (Effect Size (d)) має свою градацію в таких межах: до 0,1 — від–сутність переважаючого клінічного –ефекту; 0,1–0,4 — слабкий переважаючий ефект (Small Effect); 0,41–0,7 — середній ефект (Intermediate Effect); 0,8 і вище — великий (значний) клінічний ефект (Large Effect).
Порівняльний аналіз результатів лікування показав суттєве перевищення досягнутих клінічних ефектів в основній групі (Біірін) порівняно з контрольною: Effect Size (TGF-β1) = 1,187–1,408 та Effect Size (MMП-1) = 10,59–13,34. Відповідно, на кожних 100 пролікованих хворих практично в усіх 100 % (95% ДІ: 88,7–100,0) буде виявлено переважаючу клінічну ефективність порівняно з контрольною групою за показником MMП-1, а за показником TGF-β1 — у 68,1 % (95% ДІ: 51,4–81,8) серед хворих з вірусним гепатитом і 59,9 % (95% ДІ: 42,4–77,4) серед хворих з алкогольним гепатитом.
За іншими клінічними параметрами розмір стандартизованого переважання величини клінічного ефекту основної групи був дещо меншим, ніж за показниками MMП-1 і TGF-β1, проте виявлявся для обох груп хворих з вірусним і алкогольним гепатитом. Цілком передбачувано, що, стимулюючи експресію TGF-β1, Біірін реалізує природні антифібротичні ефекти. Посилення активності MMП-1 у групах пацієнтів, які приймали препарат Біірін, свідчить про те, що в тканинах печінки процеси руйнування й інволюції фібротичних волокон переважають над процесами фіброгенезу, що забезпечується системою матриксних металопротеїназ.
Клінічний випадок
Пацієнт С., 46 років, страждав від хронічного гепатиту С, генотип 1b. Захворювання виявили випадково. Під час одного з профілактичних обстежень у нього виявили підвищення активності АлАТ, АсАТ до 200 МО/мл. З появою пегільованих інтерферонів він успішно пройшов курс лікування інтерфероном альфа-2а у поєднанні з рибавірином протягом 11 тижнів. Лікування було успішним, вірус в крові не виділявся протягом усього контрольного періоду. При цьому періодично відзначалося підвищення активності трансаміназ (АлАТ, АсАТ), повторні дослідження на вірус гепатиту С у полімеразній ланцюговій реакції дали негативний результат. У квітні 2016 року, через понад рік після закінчення противірусної терапії, пацієнт погодився на проведення пункційної біопсії печінки. 23.04.2016 йому зробили пункційну біопсію печінки, пунктат досліджували в медичному центрі CSD Health Care 23.04.2016. Діагноз при направленні: хронічний гепатит. Цироз печінки?
Патогістологічне дослідження № 16CSD16021.
У препараті виявлено тканину печінки зі значними порушеннями архітектоніки за рахунок наявності сполучнотканинних септ між часточками. Також є ознаки формування портоцентральних септ. Зустрічаються зрідка поодинокі лімфоцити. Деякі гепатоцити з ознаками дистрофічних змін і псевдовключеннями в ядрах. Отже, у печінці є ознаки цирозу, найбільш імовірно, внаслідок перенесеного хронічного гепатиту. Індекс гістологічної активності процесу за модифікацією шкали Knodell мінімальний — 1 бал. Після біопсії протягом 3 тижнів пацієнт приймав гепатопротектори, 2 рази досліджував кров на активність печінкових ферментів і білірубін, показники білірубіну в крові коливалися від 34,8 мкмоль/л до 24,8 мкмоль/л, активність АлАТ і АсАТ — від 234 і 138 МО/мл до 132 і 93 МО/мл відповідно.
16.05.2016 пацієнт звернувся для можливого уточнення діагнозу в медичний діагностичний центр, де йому зробили еластометрію печінки, допплерографію судин черевної порожнини й ультразвукове дослідження органів черевної порожнини. Печінка збільшена незначно: ліва частка 75 мм, права частка 155 мм. Паренхіма зерниста, з дрібними рідкими ущільненнями. Ехогенність нормальна, згасання ультра–звуку в нормі. Еластометрія печінки: права частка: 10,4; 9,5; 12,3 кПа; ліва частка: 10,4; 9,6; 9,0 кПа. Загальна печінкова протока не розширена, V.portae в воротах печінки злегка розширена — 14 мм, кровотік гепато–петальний, монофазний — 20,8 см/с, у нормі. Arteria hepatis propria D = 5 мм, опір підвищено, пікова систолічна швидкість (PSV) 43,0 см/с, кінцева діастолічна швидкість (EDV) 13,3 см/с. R1 = 0,60. V.lienalis у воротах селезінки — 8 мм, кровотік сплено–фугальний — 26,2 см/с, у нормі, a.lienalis 5 мм, лінійна швидкість кровотоку збільшена, PSV = 107,5 см/с, EDV = 48,2 см/с, R1 = 0,55, селезінка розміром 115 на 53 мм.
Висновок: хронічний гепатит F3 за даними еластографії, незначно збільшена селезінка.
Після цього пацієнт звернувся по медичну допомогу до клініки, і йому було рекомендовано препарат Біірін. Прийом препарату почали з 19 травня 2016 року. Препарат застосовувався по 1 капс. 3 рази на день протягом 50 днів. Контрольні дослідження були проведені 16.09.2016. Загальний аналіз крові, біохімічні показники функції печінки в нормі. УЗД печінки й еластометрія: печінка збільшена незначно: ліва частка 75 мм і права частка 161 мм. Паренхіма зерниста, з дрібними рідкими ущільненнями. Ехогенність нормальна, згасання ультразвуку в нормі. ЕСС: жорсткість у II–III сегментах 9,0–11,0 кПa; у IV–VII сегментах — 5,0–6,0 кПa. Загальна печінкова протока не розширена, V.portae в воротах печінки злегка розширена — 12 мм. V.lienalis в воротах селезінки — 7 мм, селезінка розміром 113 на 40 мм. Жорсткість паренхіми селезінки 22–25 кПa. Висновок: реактивні зміни в печінці, F0 за шкалою METAVIR, у лівій частці печінки жорсткість паренхіми підвищена до F2, помірно виражений фіброз селезінки.
З урахуванням отриманих результатів вирішено було призначити ще один курс Бііріну. Контрольні дослідження були проведені 08.11.2016, через 2 місяці після початку другого курсу лікування. Загальний аналіз крові без змін, дослідження крові на печінкові маркери: загальний білірубін 14,2 мкмоль/л/год, АлАТ 27 МО/мл, АсАТ 19 МО/мл, тимолова проба 3,4 од. УЗД і еластометрія: розміри печінки: ліва частка 79 мм, права частка 151 мм. Паренхіма зерниста, неущільнена, ехогенність у нормі, згасання ультразвуку не підвищено. ЕСС: жорсткість у II–III сегментах 3,5–5,5 кПa; у IV–V сегментах — 5,5–6,5 кПa; у VI–VII сегментах — 6,0–7,5 кПa. V.portae у воротах печінки — 12 мм, v.lienalis у воротах селезінки — 7 мм. Селезінка розміром 114 на 38 мм, паренхіма зерниста, щільна. Жорсткість паренхіми селезінки незначно підвищена — 14–17 кПa.
Висновок: фіброз печінки F0-F1, фіброз селезінки помірно виражений.
Висновки
Результати даної роботи свідчать, що застосування препарату Біірін супроводжувалося не тільки нормалізацією показників трансаміназ, але й вірогідним зниженням рівня TGF-β1, а також підвищенням рівня MMП-1 у крові.
Отримані результати підтверджують, що комплекс рослинних пептидів і наночастинок металів — імунотропний препарат Біірін пригнічує тригерні механізми формування фіброзної тканини в печінці й сприяє дезінтеграції вогнищ фіброзу. Але найголовніше, що антифібротична дія Бііріну підтверджена даними еластографії печінки. На тлі комплексного лікування ми спостерігали не тільки поступовий регрес клінічної симптоматики захворювання, але й покращення загального стану та якості життя хворих.
Підтверджена антифібротична ефективність Бііріну дозволяє рекомендувати його як перспективний препарат для включення в схему менеджменту пацієнтів із фіброзом і цирозом печінки. Клінічні результати підтверджують, що Біірін запускає природні механізми регресу фіброзу печінки, забезпечує нормалізацію функцій і регенерацію гепатоцитів.
Список литературы
1. Guillot A, Gasmi I, Brouillet A, Ait-Ahmed Y, Calderaro J, Ruiz I, Gao B, Lotersztajn S, Pawlotsky J-M, Lafdil F. Interleukins-17 and 27 promote liver regeneration by sequentially inducing progenitor cell expansion and differentiation. Hepatol Commun. 2018 Mar;2(3):329-343.
2. Albillos A, Lario M, Alvarez-Mon M. Cirrhosis-associated immune dysfunction: distinctive features and clinical relevance. J Hepatol. 2014;61:1385-1396.
3. Alzaid F, Lagadec F, Albuquerque M, Ballaire R, Orlia–guet L, Hainault I et al. IRF5 governs liver macrophage activation that promotes hepatic fibrosis in mice and humans. JCI. 2016;Insight 1:e88689.
4. Augustin S, Millán L, González A et al. Detection of early portal hypertension with routine data and liver stiffness in patients with asymptomatic liver disease: a prospective study. J Hepatol. 2014;60(3):561-569.
5. Bansal R, van Baarlen J, Storm G, Prakash J. The interplay of the notch signaling in hepatic stellate cells and macrophages determines the fate of liver fibrogenesis. Sci. Rep. 2015;5:18272.
6. Beljaars L, Schippers M, Reker-Smit C, Martinez FO, Helming L, Poelstra K et al. Hepatic localization of macrophage phenotypes during fibrogenesis and resolution of fibrosis in mice and humans. Front. Immunol. 2014;5:430.
7. Berzigotti A, Seijo S, Arena U et al. Elastography, spleen size, and platelet count identify portal hypertension in patients with compensated cirrhosis. Gastroenterology. 2013;144(1):102-111.e1.
8. Bielen R, Moreno C, Van Vlierberghe H, Bourgeois S, Mulkay JP, Vanwolleghem T et al. The risk of early occurrence and recurrence of hepatocellular carcinoma in hepatitis C-infected patients treated with direct-acting antivirals with and without pegylated interferon: A Belgian experience. J Viral Hepat. 2017;24:976-981.
9. Bolognesi M, Di Pascoli M, Sacerdoti D. Clinical role of non-invasive assessment of portal hypertension. World J Gastroenterol. 2017;23(1):1-10.
10. Czaja AJ. Review article: the prevention and reversal of hepatic fibrosis in autoimmune hepatitis. Aliment Pharmacol Ther. 2014;39:385-406.
11. Cheng Chi, Xiao-ya Liu, Fei Hou, Xiao-zheng Yu, Chun-yun Li, Li-jian Cui, Rui-xia Liu, Cheng-hong Yin. Herbal compound 861 prevents hepatic fibrosis by inhibiting the TGF-β1/Smad/SnoN pathway in bile duct-ligated rats. BMC Complement Altern Med. 2018;18:52.
12. Colecchia A, Colli A, Casazza G et al. Spleen stiffness measurement can predict clinical complications in compensated HCV-related cirrhosis: a prospective study. J Hepatol. 2014;60(6):1158-1164.
13. Colecchia A, Montrone L, Scaioli E et al. Measurement of spleen stiffness to evaluate portal hypertension and the presence of esophageal varices in patients with HCV-related cirrhosis. Gastroenterology. 2012;143(3):646-654.
14. Ding D, Chen LL, Zhai YZ, Hou CJ, Tao LL, Lu SH, Wu J, Liu XP. Trichostatin A inhibits the activation of Hepatic stellate cells by Increasing C/EBP-α Acetylation in vivo and in vitro. Sci Rep. 2018 Mar 13;8(1):4395.
15. Dong J, Zhang X, Zhang L, Bian HX, Xu N, Bao B et al. Quercetin reduces obesity-associated ATM infiltration and inflammation in mice: a mechanism including AMPKalpha1/SIRT1. J Lipid Res. 2014;55:363-374.
16. Elkrief L, Rautou PE, Ronot M et al. Prospective comparison of spleen and liver stiffness by using shear-wave and transient elastography for detection of portal hypertension in cirrhosis. Radiology. 2015;275(2):589-598.
17. Eun HS, Jeong WI. Dual notch signaling in proinflammatory macrophage activation. Hepatology. 2016;63:1381-1383.
18. Iwakiri Y. Nitric oxide in liver fibrosis: the role of inducible nitric oxide synthase. Clin Mol Hepatol. 2015;21:319-325.
19. Kang MC, Kang SM, Ahn G et al. Protective effect of a marine polyphenol, dieckol against carbon tetrachloride-induced acute liver damage in mouse. Environmental Toxicology and Pharmacology. 2013;35(3):517-523.
20. Kim DW, Cho HI, Kim KM et al. Isorhamnetin-3-O-galactoside protects against CCl4-induced hepatic injury in mice. Biomolecules & Therapeutics. 2012;20(4):406-412.
21. Kim YJ, Park W Anti-Inflammatory effect of quercetin on RAW 264.7 mouse macrophages induced with polyinosinic-polycytidylic acid. Molecules. 2016;21:450.
22. Kruse RL, Kramer JR, Tyson GL, Duan Z, Chen L, El-Serag HB et al. Clinical outcomes of hepatitis B virus coinfection in a United States cohort of hepatitis C virus-infected patients. Hepatology. 2014;60:1871-1878.
23. Lassailly G et al. Bariatric surgery reduces features of nonalcoholic steatohepatitis in morbidly obese patients. Gastroentero––logy. 2015;149(2):379-388.
24. Ling Lan, Ran Liu, Ling-Yun Qin, Peng Cheng, Bo-Wei Liu, Bing-Yong Zhang, Song-Ze Ding, Xiu-Ling Li. Transplantation of bone marrow-derived endothelial progenitor cells and hepatocyte stem cells from liver fibrosis rats ameliorates liver fibrosis. World J Gastroenterol. 2018 Jan 14;24(2):237-247.
25. Loomba R, Cui J, Wolfson T et al. Novel 3D magnetic resonance elastography for the noninvasive diagnosis of advanced fibrosis in NAFLD: a prospective study. Am J Gastroenterol. 2016;111(7):986-994.
26. Loomba R, Wolfson T, Ang B et al. Magnetic resonance elastography predicts advanced fibrosis in patients with nonalcoholic fatty liver disease: a prospective study. Hepatology. 2014;60(6):1920-1928.
27. Manns M, Samuel D, Gane EJ et al. Ledipasvir and sofosbuvir plus ribavirin in patients with genotype 1 or 4 hepatitis C virus infection and advanced liver disease: a multicentre, open-label, randomised, phase 2 trial. Lancet Infect Dis. 2016;16(6):685-697.
28. Mattar EH, Almehdar HA, Uversky VN, Redwan EM. Virucidal activity of human α- and β-defensins against hepatitis C virus genotype 4. Mol Biosyst. 2016 Aug 16;12(9):2785-97.
29. Na Li, Jinlian Hua. Immune cells in liver regeneration. Oncotarget. 2017 Jan 10;8(2):3628-3639.
30. Petruzziello A, Marigliano S, Loquercio G, Cozzolino A, Cacciapuoti C. Global epidemiology of hepatitis C virus infection: An up-date of the distribution and circulation of hepatitis C virus genotypes. World J Gastroenterol. 2016;22:7824-7840.
31. Peverill W, Powell LW, Skoien R. Evolving concepts in the pathogenesis of NASH: beyond steatosis and inflammation. Int J Mol Sci. 2014;15:8591-8638.
32. Halota W, Flisiak R, Juszczyk J, Małkowski P, Pawłow–ska M et al, Polish Group of Experts for HCV. Recommendations for the treatment of hepatitis C in 2017. Clin Exp Hepatol. 2017;3:47-55
33. Prenner SB, VanWagner LB, Flamm SL, Salem R, Lewandowski RJ, Kulik L. Hepatocellular carcinoma decreases the chance of successful hepatitis C virus therapy with direct-acting antivirals. J Hepatol. 2017;66:1173-1181.
34. Raff EJ, Kakati D, Bloomer JR, Shoreibah M, Rasheed K, Singal AK. Diabetes mellitus predicts occurrence of cirrhosis and hepatocellular cancer in alcoholic liver and non-alcoholic fatty liver diseases. J Clin Transl Hepatol. 2015;3:9-16.
35. Rao H, Wu E, Fu S, Yang M, Feng B, Lin A et al. The higher prevalence of truncal obesity and diabetes in American than Chinese patients with chronic hepatitis C might contribute to more rapid progression to advanced liver disease. Aliment Pharmacol Ther. 2017;46:731-740.
36. Reddy KR, Bourliere M, Sulkowski M et al. Ledipasvir and sofosbuvir in patients with genotype 1 hepatitis C virus infection and compensated cirrhosis: An integrated safety and efficacy analysis. Hepatology. 2015;62(5):79-86.
37. Rehm J, Samokhvalov AV, Shield KD. Global burden of alcoholic liver diseases. J Hepatol. 2013;59:160-168.
38. Song X, Liu W, Xie S, Wang M, Cao G, Mao C et al. All-transretinoic acid ameliorates bleomycin-induced lung fibrosis by downregulating the TGF-beta1/Smad3 signaling pathway in rats. Lab. Invest. 2013;93:1219-1231.
39. Secchi MF, Crescenzi M, Masola V, Russo FP, Floreani A, Onisto M. Heparanase and macrophage interplay in the onset of liver fibrosis. Scientific Reports. 2017;7:14956.
40. Singal AG, Pillai A, Tiro J. Early detection, curative treatment, and survival rates for hepatocellular carcinoma surveillance in patients with cirrhosis: a meta-analysis. PLoS Med. 2014;11(4):e1001624.
41. Tachi Y, Hirai T, Toyoda H et al. Predictive abili–ty of laboratory indices for liver fibrosis in patients with chronic hepatitis C after the eradication of hepatitis C virus. PLoS One. 2015;10(7):e0133515.
42. Tacke F. Targeting hepatic macrophages to treat liver di–seases. J Hepatol. 2017 Jun;66(6):1300-1312.
43. Takashi Matsuda, Taro Takami, Ryo Sasaki, Tatsuro Nishimura, Yuki Aibe, Bruno Diaz Paredes, Luiz Fernando Quintanilha, Toshihiko Matsumoto, Tsuyoshi Ishikawa, Naoki Yamamoto, Kenji Tani, Shuji Terai, Yasuho Taura, Isao Sakaida. A canine liver fibrosis model to develop a therapy for liver cirrhosis using cultured bone marrow-derived cells. Hepatol Commun. 2017 Sep;1(7):691-703.
44. Kisseleva T, Cong M, Paik YH, Scholten D, Jiang C, Benner C, Iwaisako K, Moore-Morris T, Scott B, Tsukamoto H, Evans SM, Dillmann W, Glass CK, Brenner DA. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc Natl Acad Sci U S A. 2012 Jun 12;109(24):9448-9453.
45. Tosello-Trampont AC, Krueger P, Narayanan S, Lan–des SG, Leitinger N, Hahn YS. NKp46(+) natural killer cells attenuate metabolism-induced hepatic fibrosis by regulating macrophage activation in mice. Hepatology. 2016;63:799-812.
46. Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. 2017;14:397-411.
47. Tung-Hung S, Jia-Horng K, Chun-Jen L. Molecular Mechanism and Treatment of Viral Hepatitis-Related Liver Fibrosis. Int J Mol Sci. 2014 Jun;15(6):10578-10604.
48. Wan J, Benkdane M, Teixeira-Clerc F, Bonnafous S, Louvet A, Lafdil F et al. M2 kupffer cells promote M1 kupffer cell apoptosis: a protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology. 2014;59:130-142.
49. Waziry R, Hajarizadeh B, Grebely J, Amin J, Law M, Danta M et al. Hepatocellular carcinoma risk following direct-acting antiviral HCV therapy: A systematic review, meta-analyses, and meta-regression. J Hepatol. 2017;67:1204-1212.
50. Weng SY, Wang X, Vijayan S, Tang Y, Kim YO, Padberg K, Regen T, Molokanova O, Chen T, Bopp T, Schild H, Brombacher F, Crosby JR, McCaleb ML, Waisman A, Bockamp E, Schuppan D. IL-4 Receptor Alpha Signaling through Macrophages Differentially Regulates Liver Fibrosis Progression and Reversal. EBioMedicine. 2018 Mar;29:92-103.
51. Wijesundera KK, Izawa T, Tennakoon AH, Murakami H, Golbar HM, Katou-Ichikawa C et al. M1- and M2-macrophage polarization in rat liver cirrhosis induced by thioacetamide (TAA), focusing on Iba1 and galectin-3. Exp. Mol. Pathol. 2014;96:382-392.
52. Wilhelm A, Aldridge V, Haldar D, Naylor AJ, Weston CJ, Hedegaard D et al. CD248/endosialin critically regulates hepatic stellate cell proliferation during chronic liver injury via a PDGF-regulated mechanism. Gut. 2016;65:1175-1185.
53. Wu L, Zhang Q, Mo W, Feng J, Li S, Li J et al. Quercetin prevents hepatic fibrosis by inhibiting hepatic stellate cell activation and reducing autophagy via the TGF-β1/Smads and PI3K/Akt pathways. Sci Rep. 2017;7:9289.
54. Wynn TA, Chawla A, Pollard JW. Macrophage bio–logy in development, homeostasis and disease. Nature. 2013;496:445-455.
55. Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. 2016;44:450-462.
56. Young Kul Jung, Hyung Joon Yim. Reversal of liver cirrhosis: current evidence and expectations. Korean J Intern Med. 2017 Mar;32(2):213-228.
57. Yue-Ming Ling, Jin-Yu Chen, Libin Guo, Chen-Yi Wang, Wen-Ting Tan, Qing Wen, Shu-Dong Zhang, Guo-Hong Deng, Yao Lin, Hang Fai Kwok. β-defensin 1 expression in HCV infected liver/liver cancer: an important role in protecting HCV progression and liver cancer development. Sci Rep. 2017;7:13404. Published online 2017 Oct 17.
58. Zhong W, Qian K, Xiong J, Ma K, Wang A, Zou Y. Curcumin alleviates lipopolysaccharide induced sepsis and liver failu–re by suppression of oxidative stress-related inflammation via PI3K/AKT and NF-κB related signaling. Biomedicine & Pharmacotherapy. 2016;83:302-313.
59. Zaulet M, Kevorkian SEM, Dinescu S et al. Protective effects of silymarin against bisphenol A-induced hepatotoxi–city in mouse liver. Experimental and Therapeutic Medicine. 2017;13(3):821-828.

/u/7-1.jpg)
/u/8-1.jpg)