Вступ
Нервово-м’язові захворювання (НМЗ) — група гетерогенних захворювань із різними клінічними проявами, головними з яких є прогресуюча м’язова слабкість і стомлюваність, різний вік дебюту, підвищений рівень ферменту креатинфосфокінази (КФК) і дистрофічні зміни м’язових волокон.
НМЗ є одними з найпоширеніших серед спадкових хвороб. У 1991 році виконано оглядове дослідження нервово-м’язових порушень, опубліковане в рецензованій літературі. З того часу діагностика значно покращилася завдяки генетичному підтвердженню і розробленим діагностичним критеріям. У період із 1990 по 2014 р. PubMed проведено дослідження епідеміології, захворюваності та поширеності щодо тридцяти НМЗ для рецензованої літератури. Були виявлені показники захворюваності для десяти розладів у діапазоні від 0,05 до 9 на 100 000 на рік. Показники поширеності більшості НМЗ від 1 до 10 на 100 000 населення, за винятком мультифокальної рухової невропатії, міастенічного синдрому Ламберта — Ітона, дистрофії Емері — Дрейфуса, окулофарингеальної м’язової дистрофії та вроджених м’язових дистрофій, які становили ± 1/100 000. Для постполіомієлітного синдрому і хвороби Шарко — Марі — Тута виявили поширеність ± 10/100 000.
На першому десятилітті життя дебютує більшість клінічних форм прогресуючих м’язових дистрофій й аміотрофій, що мають прогредієнтний перебіг. Останнім десятиліттям досягнуті значні успіхи у вивченні молекулярних механізмів спадкових нейром’язових захворювань. Практично при всіх формах нейром’язової патології виявлені конкретні мутантні гени на хромосомах. При багатьох формах встановлений продукт гена — білок, що контролює мутантний ген, який є причиною первинного біохімічного дефекту. На підставі світових даних, новітніх розробок у галузі молекулярної діагностики, превентивної діагностики та генної терапії в журналі «Neuromuscular Disorders» щорічно в грудневому випуску публікується оновлений список моногенних м’язових захворювань внаслідок первинного дефекту, що відмічається в ядерному геномі. Він включає хвороби, в яких причинний ген відомий або принаймні локалізований на хромосомі, якщо він ще не ідентифікований. Захворювання, для яких локус не нанесений на карту або які пов’язані з дефектами, пов’язаними з мітохондріальними генами, не включені.
На сьогодні розроблені діагностичні критерії основних форм спадкових НМЗ, вивчені окремі ланки їх патогенезу. Однак виражена клініко-генетична гетерогенність НМЗ обумовлює значну складність для їх диференціальної діагностики.
Протягом останніх десятиліть магнітно-резонансна візуалізація (МРТ) стала вирішальною для діагностики захворювань м’яких тканин. Діагностична цінність МРТ була переконливо доведена при спадкових і запальних нервово-м’язових захворюваннях. На додаток до неврологічного та нейрофізіологічного дослідження, нейром’язова МРТ є цінним діагностичним інструментом, що дозволяє оцінити ступінь і, що більш важливо, участь м’язів у патологічному процесі.
Сьогодні магнітно-резонансна томографія є стандартним методом, що допомагає в діагностиці спадкових нервово-м’язових захворювань, а також диференціювати ряд м’язової дистрофії. МРТ не тільки є діагностичним методом, а й відіграє важливу роль при плануванні біопсії, є методом оцінки результатів лікування.
МРТ м’язів — неінвазивний метод, що дає змогу виявити дегенеративні зміни, жирову інфільтрацію, наявність набряку і запальних змін скелетної мускулатури. Ця методика також досить чітко дозволяє розмежувати анатомічні структури досліджуваної ділянки: виділити окремі м’язи, судинно-нервові пучки, підшкірно-жирову клітковину, фасції, кістки.
МРТ — метод, заснований на отриманні серій зображень у фронтальній й аксіальній площині зрізів із використанням певних імпульсних послідовностей. Інтенсивність сигналу тканини скелетних м’язів при T1-зважених зображеннях трохи вища, ніж у води, і значно нижча, ніж у жиру. На T2-зваженому зображенні інтенсивність сигналу м’язової тканини набагато нижча, ніж води або жирової тканини. Через короткий час релаксації T1-сигналу інтенсивність жирової тканини є яскравою на T1-зваженій МРТ. Жир також показує високу інтенсивність сигналу на T2-зважених зображеннях. З огляду на те, що і жирові тканини, і вода на Т2-імпульсній послідовності виглядають світлими, неможливо відрізнити жир від набряку лише на підставі серії Т2-імпульсних послідовностей. Простий спосіб відрізнити жир і воду на T2-зважених зображеннях — застосовувати жироподавлення. При подавленні жиру (Fat Sat) за Т2-зваженими зображеннями або в STIR-режимі сканування інтенсивність м’язового сигналу нижча, ніж води, і трохи нижча, ніж у жировій тканині. Інакше кажучи, у цьому разі імпульсна послідовність Т2-FS/STIR видалить сигнал від жирових тканин, що дозволить розрізнити набряк і жирову дегенерацію. Особливо важлива ця послідовність в окремих випадках, коли є підозри на запальні зміни в м’язовій тканині. STIR-імпульсна послідовність може бути використана замість Т2-імпульсної послідовності. У низці випадків замість STIR-імпульсної послідовності використовують PD-імпульсну послідовність (зважена за протонною щільністю), у тому числі в поєднанні з придушенням сигналу від жиру, що також дозволяє візуалізувати набряк.
Для аналізу зображень найбільш підходять такі типи тканин — м’язи і жир. Аналіз зображення м’язової тканини в пацієнтів із підозрою на певні нервово-м’язові захворювання повинен включати такі параметри: форму (нормальна конфігурація, деформація); розмір (нормальний, атрофічний і гіпертрофічний); тканинну архітектуру (однорідна, ознаки дегенерації, сполучна тканина); вогнищеві та об’ємні ураження (кальцифікація, змішані ураження тканини); аномалії сигналів (набряк і запалення). Перші три аспекти аналізу зображень повинні виконуватися на T1-зваженій візуалізації. Форма і розмір м’язів розрізняються серед індивідуумів залежно від декількох факторів (наприклад, харчування, фізичні вправи, вік, стать). Нормальна тканинна архітектура скелетних м’язів відрізняється серед м’язових груп й анатомічних місць.
Існують рейтингові шкали МРТ щодо візуальної оцінки дистрофічних змін (жирова дегенерація) смугастої м’язової тканини. У практичній роботі частіше використовується класифікація Mercuri (2002) (табл. 1), що становить собою адаптовану МРТ-версію класифікації томоденситометрії, запропонованої Goutallier і Bernageau, що дозволяє оцінити ступінь дегенерації м’язів плеча. Так само використовуються й шкали Kornblum et al. (2006) і Fischer et al. (2008) (табл. 1).
Існує 5-бальна шкала, запропонована А. Lammi-nen (1990) у модифікації Н. Jungbluth et al. (2004), для оцінки структури і патологічних змін інтенсивності сигналу м’язів нижніх кінцівок (табл. 2).
Нижче ми наводимо клінічний приклад, у діагностиці якого була використана МРТ м’язів.
Клінічний приклад
Хвора Зоряна Г., 30 років, надійшла в клініку Обласного клінічного центру неврології та нейрохірургії м. Ужгорода у плановому порядку зі скаргами на виражену слабкість і відчуття стягування в ногах, труднощі при ходьбі, значне утруднення при підйомі сходами, неможливість перейти з положення сидячи в положення стоячи без сторонньої допомоги, труднощі вставання зі стільця, зміну ходи — ходить, як каченя, труднощі при піднятті рук догори.
З анамнезу відомо, що вважає себе хворою протягом 10 років, коли після перших пологів стала відчувати епізоди слабкості в ногах, в подальшому слабкість стала постійною. Зверталася до невролога за місцем проживання, отримувала терапію з приводу остеохондрозу поперекового відділу хребта. Близько 5 років тому відзначала незрозумілий епізод лихоманки (до 39 °С) протягом 2–3 діб із вираженою загальною слабкістю, що регресувала на тлі прийому нестероїдних протизапальних засобів. Згодом зазначає наростання слабкості, з’явилися болі в нижніх кінцівках, відчуття стягування в них, стало тяжко ходити (останні 3 роки), вставати зі стільця, виникла проблема при підйомі сходами, при піднятті рук вгору, особливо при розчісуванні, останнім часом змінилася хода — виглядає як качина. Звернулася на амбулаторний прийом Обласного клінічного центру нейрохірургії та неврології (ОКЦНН) м. Ужгорода, де була виконана електронейроміографія, на якій виявлені ознаки генералізованого, міопатичного типу ураження з переважанням у нижніх кінцівках проксимально (хвороба накопичення?).
Анамнез життя: друга вагітність закінчилася смертю плода.
Неврологічний статус при надходженні: свідомість за шкалою коми Глазго — 15 балів. Контакту доступна. Мовлення збережене, чітке. Очні щілини D = S, зіниці D = S. Обличчя симетричне, язик розташований медіально. Ковтання не порушене. Глотковий рефлекс збережений. Рухливість м’якого піднебіння при фонації збережена. Сухожилкові рефлекси з рук, ахіллові і колінні відсутні. Патологічні кистьові знаки відсутні. Тонус м’язів нижніх кінцівок знижений. Атрофії, гіпотрофії не виявлено. М’язова сила в нижніх кінцівках знижена до 3 балів (проксимально), в руках — до 4 балів (проксимально). Чутливих розладів не показує. У позі Ромберга стійка. Стояння на носках утруднене, на п’ятах — неможливе. Позитивний симптом Говерса. ПНП виконує. Тазові функції контролює.
Додаткові обстеження в умовах ОКЦНН: загальноклінічні аналізи: в нормі. РВ: негативне. IgG до Borrelia burgdorferi не виявлено. Клінічна хімія: креатинін — 39 ммоль/л, аланінамінотрансфераза — 52, аспартатамінотрансфераза — 47, креатинфосфокіназа — 929, тиреотропний гормон, Т4, Т3 — у нормі. Ревмопроби, лактат у нормі. ANA-комплекс, ANA, метод IFT — негативний результат. ЕхоКГ без патологічних змін. Rö-ОГК у нормі. Генетичний аналіз альфа-галактозидази негативний. МРТ поперекового відділу хребта: гіперлордоз, периневральна кіста Тарлова S2-рівня. Від обстеження в медико-генетичному центрі і м’язової біопсії, на жаль, пацієнт відмовився.
Диференціальна діагностика проводилася з низкою захворювань, з урахуванням насамперед ознак прогресуючої симетричної проксимальної м’язової слабкості кінцівок, що характеризуються розвитком аналогічних симптомів у різних комбінаціях: ендокринної міопатії, хвороби Помпе, прогресуючої м’язової дистрофії, дистрофінопатії Беккера, тазово-стегнової міодистрофії Лейдена — Мебіуса, кінцівково-поясної м’язової дистрофії, спінальної м’язової атрофії Олбрайта і запальних міопатій (дермато-/поліміозит).
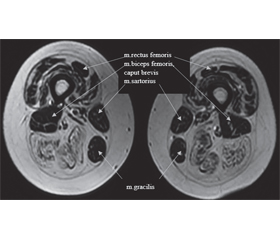
Був додатково досліджений міозитний профіль, виявлені антитіла до треоніл-тРНК-синтетази, PL-7. Виконана МРТ м’язів стегон і тазового пояса (сканер General Electric, Signa HDxt 1.5T, ЗДЦ «Євроклінік», м. Ужгород): відзначається симетрична білатеральна жирова дегенерація окремих м’язів стегон: субтотальне заміщення жировою тканиною волокон м’язів: m. vastus lateralis, m. vastus medialis, m. vastus intermedius, m. adductor magnus, m. adductor longus, що за ступенем тяжкості трохи переважає з правого боку, і повне двостороннє заміщення жировою тканиною м’язів: m. semitendinosus, m. semimembranosus, m. biceps femoris: caput longus, m.quadratus femoris, м’язи мають нерівні нечіткі контури, узурповано за типом тканини, «поїденою міллю», із множинними різнокаліберними ділянками підвищеного сигналу, які займають від 20 до 80 % їх обсягу — поліморфна картина з ураженням м’язів від початкового до вираженого (1-ша — 3-тя стадії за шкалами Mercuri і Fischer, 1-ша — 2-га стадії за шкалою Kornblum). Крім того, запальні зміни, такі як набряк м’язової тканини, також добре візуалізуються при МРТ у STIR-послідовності (рис. 1, 2). Висновок: МР-ознаки жирової дистрофії м’язів тазової ділянки і стегон.
Консультована ревматологом: поліміозит (антисинтетазний синдром).
У відділенні отримувала терапію: від запропонованої терапії ГКС хвора відмовилася, у зв’язку з чим отримувала кокарніт, корвітин, тіогама турбо, цитофлавін, нейромідин, біовен моно, на тлі чого відзначала: регрес болю в ногах, збільшилась м’язова сила в кінцівках (може піднятися на 2–3-й поверх), зменшилося відчуття стягування в ногах, може пройти до 500 м без відпочинку.
З огляду на клінічну картину, повільно прогресуючий тип перебігу, відсутність екстраневральної патології, дані додаткових досліджень пацієнтці встановлений діагноз: кінцівково-поясна м’язова дистрофія з тетрапарезом (легким у руках, помірним у ногах, порушеннями функції ходьби).
Припущення комбінації міопатії з поліміозитом не може бути відкинуте, незважаючи на тривалий перебіг захворювання та відсутність екстраневральної симптоматики, у зв’язку з наявністю антитіл до треоніл-тРНК-синтетази, PL-7, що зобов’язує нас продовжити діагностичний пошук.
Існують окремі повідомлення про пацієнток жіночої статі з ознаками міопатії, які імітували запальний міозит як прояв носійства гена міодистрофії Дюшена. Клініцист повинен проявляти обережність при діагностиці запального міозиту на основі клінічних критеріїв, оскільки м’язова дистрофія в пацієнтів старшого віку може проявлятися запальними ознаками. У цьому випадку головне місце в діагностиці належить генетичному консультуванню для встановлення правильного діагнозу.
Висновки
Таким чином, застосування МРТ м’язів для діагностики НМЗ є новим перспективним методом, що дозволяє значно спростити диференціально-діагностичний пошук. МРТ м’язів нерідко відіграє вирішальну роль у постановці діагнозу. З огляду на наявні дані можна стверджувати, що МРТ м’язів — високоінформативний метод для хворих із нервово-м’язовою патологією, що може застосовуватися не тільки з діагностичною метою, але і для оцінки ступеня дегенерації м’язової тканини в динаміці.
Конфлікт інтересів. Автори заявляють про відсутність конфлікту інтересів при підготовці даної статті.
Список литературы
1. Евтушенко С.К., Евтушенко О.С., Евтушенко Л.Ф., Евтушенко И.С. Ранняя клинико-инструментальная диагностика и терапия быстро- и медленно прогрессирующих мышечных дистрофий и амиотрофий // Международный неврологический журнал. — 2007. — № 4(14). — С. 52-64.
2. Евтушенко С.К., Евтушенко О.С., Евтушенко И.С., Шаймурзин М.Р. Нейромышечные заболевания. — К.: Ноулидж, 2014. — 215 с.
3. Пальшина С.Г., Никитин С.С., Васильев В.И. Опыт применения ритуксимаба в лечении полимиозита с антисинтетазным синдромом // Нервно-мышечные болезни. — 2012. — 3. — С. 45-54.
4. Антелава О.А., Раденска-Лоповок С.Г., Насонов Е.Л. Диагностические критерии идиопатических воспалительных миопатий. Проблемы их оптимизации // Современная ревматология. — 2014. — 3. — С. 56-65.
5. Carlier R. МРТ мышц/МРТ всего тела в диагностике и динамическом наблюдении пациентов с нервно-мышечными заболеваниями: Пер. М.О. Ковальчук // Нервно-мышечные болезни. — 2014. — 2. — С. 16-26.
6. Влодавец Д.В., Казаков Д.О. Диагностические возможности магнитно-резонансной томографии мышц при нервно-мышечных заболеваниях // Неврологический журнал. — 2014. — 3. — С. 4-12.
7. Carlier P.G., Marty B., Scheidegger O., de Sousa P.L., Baudin P.-Y., Snezhko E., Vlodavets D. Роль количественной магнитно-резонансной томографии и спектроскопии скелетных мышц в оценке результатов клинических исследований (часть I) // Нервно-мышечные болезни. — 2016. — 4. — С. 10-20.
8. Carlier P.G., Marty B., Scheidegger O., de Sousa P.L., Baudin P.-Y., Snezhko E., Vlodavets D. Роль количественной магнитно-резонансной томографии и спектроскопии скелетных мышц в оценке результатов клинических исследований (часть I) // Нервно-мышечные болезни. — 2017. — 1. — С. 11-29.
9. Шнайдер Н.А., Николаева Т.Я., Бороева Е.Н., Пшеннико-ва Г.М., Лугинов Н.В., Панина Ю.С. Конечностно-поясная мышечная дистрофия с аутосомно-доминантным типом наследования: пельвиофеморальная форма Лейдена — Мебиуса // Нервно-мышечные болезни. — 2013. — 1. — С. 46-61.
10. Егоркина О.В., Волошина Н.П. Идиопатические воспалительные миопатии // Ліки України. — 2011. — 4(150). — С. 68-76.
11. Brockmann K., Becker P., Schreiber G., Neubert K., Brunner E., Bonnemann C. Sensitivity and specifiity of muscle ultrasound in assessment of suspect neuromuscular disease in childhood // Neuromuscul. Disord. — 2007. — 17. — Р. 517-23.
12. Hobson-Webb L., Burns T.M. The more the merrier muscle and Nerve. — 2008. — 37. — Р. 555-9.
13. Yoon J., Kim S.H., Ki C.-S., Kwon M.-J., Lim M.-J., Kwon S. R., Lim M.-J., Kwon S.-R., Joo K., Moon C.-G., Park W. Carrier Woman of Duchenne Muscular Dystrophy Mimicking Inflammatory Myositis // Journal of Korean Medical Science. — 2011. — 26(4). — Р. 587.
14. Hoogerwaard E., Bakker E., Ippel P., Oosterwijk J., Majoor-Krakauer D., Leschot N., Van Essen A., Brunner H., van der Wouw P., Wilde A., de Visser M. Signs and symptoms of Duchenne muscular dystrophy and Becker muscular dystrophy among carriers in the Netherlands: a cohort study // The Lancet. — 1999. — 353(9170). — Р. 2116-2119.
15. Tasca G., Monforte M., Iannaccone E., Laschena F., Ottaviani P., Silvestri G., Masciullod M., Mirabellab M., Servideib S., Ricci E. Muscle MRI in female carriers of dystrophinopathy // European Journal of Neurology. — 2012. — 19(9). — Р. 1256-1260.
16. May D.A., Disler D.G., Jones E.A., Balkissoon A.A., Manaster B.J. Abnormal Signal Intensity in Skeletal Muscle at MR Imaging: Patterns, Pearls, and Pitfalls // RadioGraphics. — 2000. — 20 (Suppl. 1). — Р. 295-315.
17. Lovitt S., Moore S.L., Franklin A., Marden F.A. The use of MRI in the evaluation of myopathy // Clinical Neurophysiology. — 2006. — 117. — Р. 486-495.

/36.jpg)
/37.jpg)