
Международный неврологический журнал №4 (106), 2019
Вернуться к номеру
Епілептичний міоклонус повік з абсансами (синдром Дживонса): огляд літератури й клінічне спостереження
Авторы: Кирилова Л.Г. (1), Мірошников О.О. (1), Погребняк А.Б. (2), Юзва О.О. (1)
1 - ДУ «Інститут педіатрії, акушерства і гінекології імені академіка О.М. Лук’янової НАМН України», м. Київ, Україна
2 - Медичний центр «Лікарська практика», м. Київ, Україна
Рубрики: Неврология
Разделы: Справочник специалиста
Версия для печати
У статті подані сучасні уявлення щодо етіології, патогенезу, клінічних проявів, підходів до діагностики й лікування рідкісної форми ідіопатичної генералізованої епілепсії — синдрому Дживонса. Також наводиться опис клінічного випадку епілептичного міоклонусу повік з абсансами (синдрому Дживонса) у підлітка.
В статье приведены современные представления об этиологии, патогенезе, клинических проявлениях, подходах к диагностике и лечению редкой формы идииопатической генерализованной эпилепсии — синдрома Дживонса. Также представлено описание клинического случая эпилептического миоклонуса век с абсансами (синдрома Дживонса) у подростка.
The article deals with modern concepts of etiology, pathogenesis, clinical manifestations, approaches to the diagnosis and treatment of rare form of idiopathic generalized epilepsy — Jeavons syndrome. The paper also presents a case report of the eyelid myoclonia with absences (Jeavons syndrome) in teenager.
синдром Дживонса; ідіопатичні генералізовані епілепсії; міоклонус повік; абсанси; фотосенситивність; огляд
синдром Дживонса; идиопатические генерализованные эпилепсии; миоклонус век; абсансы; фотосенситивность; обзор
Jeavons syndrome; idiopathic generalized epilepsy; eyelid myoclonia; absences; photosensitivity; review
Вступ
Існують пароксизмальні стани, які не так легко своє–часно розрізнити, діагностувати та призначити адекватну терапію. Саме до такої групи захворювань відноситься синдром Дживонса (СД).
Синдром Дживонса (Jeavons syndrome) — генетично детермінована генералізована форма рефлекторної епілепсії, що характеризується початком у дитячому віці, своєрідною семіотикою епілептичних припадків, вираженою фотосенситивністю, а також можливістю виникнення генералізованих тоніко–клонічних нападів. Інша назва цього захворювання — міоклонус повік з абсансами або без абсансів [1]. Синдром Дживонса вважається рідкісним (орфанним) захворюванням, якому присвоєний номер 139431 у каталозі порталу orpha.net [2]. Захворювання вперше описано в 1977 році P.M. Jeavons, який у подальшому неодноразово уточнював клінічні прояви синдрому [3]. Однак ще в 1932 році M. Radovici описав 20–річну пацієнтку з міоклоніями повік, абсансами і вираженою фотосенситивністю, які виникли у віці 10–12 років, що дає підставу розглядати «синдром Радовічі» як потенційно можливу назву цього захворювання [4]. Надалі серед дослідників існували протиріччя, чи є синдром Дживонса окремим епілептичним синдромом або лише проявом інших ідіопатичних і криптогенних епілепсій [1, 5, 6].
В 1996 р. J.S. Duncan і C.P. Panayiotopoulos у своїй монографії запропонували називати міоклонус повік з абсансами епонімом «синдром Дживонса» на честь автора першого наукового опису цього захворювання [7]. Натомість підхід Міжнародної протиепілептичної ліги (ILAE) до класифікації СД змінювався з часом. У першій класифікації епілептичних припадків та епілепсій СД формально не визнавали як окремий синдром, проте з 2010 року класифікація епілепсій та епілептичних припадків ILAE визнає міоклонус повік як один із видів абсансних припадків, а саме абсанс з особливими властивостями (absence seizure with special features) [8]. У класифікації 2014 року міоклонус повік віднесли до одного з чотирьох окремих видів абсансних припадків [9]. У 2017 р. робоча група ILAE під керівництвом R.S. Fisher визнала міоклонус повік як один із нових генералізованих видів абсансних припадків (absence seizures, with eyelid myoclonia) [10]. На сьогодні, відповідно до рекомендацій робочої групи з класифікації епілепсії ILAE 2017 року під керівництвом Ingrid E. Scheffer, СД можна віднести до генетичної епілепсії з генералізованими немоторними припадками (абсансами) і міоклонією повік [11].
Епідеміологія
Точні дані щодо поширеності СД у дітей відсутні, але він становить 2,5–3 % всіх випадків епілепсій у дорослих. У структурі генетичних генералізованих епілепсій CД становить 7,3–12,9 % та 13 % від усіх генералізованих епілепсій з абсансами. Таким чином, поширеність СД є не меншою, ніж ювенільної міолоклонічної епілепсії, проте, на відміну від останньої, він значно рідше діагностується [5, 12].
Більшість авторів вважає, що СД частіше виникає у жінок. Співвідношення пацієнтів чоловічої та жіночої статі — 1,25–3,2 : 4,1. Проте у різних дослідженнях співвідношення між пацієнтами чоловічої та жіночої статі значно відрізнялося [5].
Клінічні прояви
Дебют СД зазвичай відбувається у 2–14 років з піком у 6–8 років. Середній вік перших проявів захворювання — 6,5 ± 2,5 року. Однак точний вік появи нападів складно встановити, оскільки часто у дітей міоклонії повік розцінюються як тики чи манеризми, а їх епілептична природа та наявність супутніх абсансів тривалий час можуть залишатися нерозпізнаними [5, 13].
Найбільш важливим симптомом СД є міоклонії повік, що можуть супроводжуватися короткочасними (3–6 секунд) абсансами або без них. Міоклонії повік можуть проявлятися у вигляді їх швидкого посмикування, тремору, тріпотіння або вібрації. Рухи повік можуть бути ритмічними або нерегулярними, одиничними або множинними, незначними або вираженими. Міоклонії повік часто супроводжуються різким відведенням очних яблук або голови вгору, але важливим діагностичним моментом є те, що при СД ніколи не відмічається відведення очей вбік. Напади найчастіше є фотосенситивними, тобто з’являються при погляді на блимаюче або пряме світло. З віком, як правило, фотосенситивність зменшується [12, 13].
Також особливістю СД є виникнення припадку після заплющування очей. Для СД характерним є феномен fixation–off sensitivity (FOS), що полягає у виникненні міоклоній повік та одночасно іктальної епілептиформної активності на електроенцефалографії (ЕЕГ) після заплющування очей та їх утримування у заплющеному стані [12, 13]. Таким чином, напади при СД провокуються прямим світлом, а також заплющуванням очей. Міоклонії повік виникають багаторазово протягом доби. У 20 % пацієнтів може розвиватися статус міоклоній повік у вигляді частих повторних припадків [12].
Відразу за міоклонією повік може виникати абсанс, який проявляється частковим порушенням свідомості з короткочасною зупинкою рухової діяльності, повторенням рухів або слів, появою помилок або затримок при рахуванні. Часто виникають латентні абсанси, що залишаються непомітними як для самого пацієнта, так і для оточуючих. Абсанси, що супроводжують міоклонії повік, зазвичай малопомітні та короткі (< 1 секунди). Абсанси при синдромі Дживонса також можуть виникати незалежно від заплющування очей під час гіпервентиляції або ритмічної фотостимуляції, у зв’язку з цим Jeavons в 1996 р. запропонував змінити назву захворювання з «міоклонус повік з абсансами» на «міоклонус повік та абсанси», підкреслюючи можливість виникнення абсансів, незалежних від міоклоній повік [14].
Іноді у пацієнта можуть виникати міоклонії шиї або верхніх кінцівок, однак вони не типові та рідкісні (за деякими даними, близько 30–50 % пацієнтів). Часто хворі відчувають лише суб’єктивне відчуття міоклоній шиї або кінцівок, але вони не помітні оточуючим [15]. Деякі автори вважають, що поява міоклоній іншої локалізації, крім повік, виключає діагноз «синдром Дживонса» [1].
Описані випадки автоіндукції припадків при СД [1]. Поява міоклоній повік після раптової дії яскравого світла часто помилково розцінюється як феномен автоіндукції припадків або як моторні тики м’язів обличчя. Також у деяких пацієнтів, які не отримують лікування, можливий розвиток звичних міоклоній повік без супутніх змін на ЕЕГ. У 50 % пацієнтів можуть виникати рідкісні генералізовані тоніко–клонічні напади, що можуть бути фотоспровокованими або спонтанними. У більшості пацієнтів тоніко–клонічні напади неодмінно приєднуються з часом та особливо схильні до виникнення після дії провокуючих факторів (депривація сну, алкоголь, втома, яскраве світло) або неадекватної антиконвульсивної терапії. Як правило, великі напади добре піддаються лікуванню антиконвульсантами. Вважається, що при синдромі Дживонса вплив припадків на когнітивні функції є мало вираженим, але описані пацієнти з помірним зниженням інтелекту. При СД може розвиватися неконвульсивний епілептичний статус, який проявляється тривалою сплутаністю свідомості. Фотостимуляція при цьому може викликати генералізовані тоніко–клонічні напади [5, 8, 13].
Виділяють такі клінічні форми синдрому Дживонса [5, 13, 15]:
1. СД з раннім початком (до 4 років):
а) типова форма — характерним проявом цієї форми є звичайне для СД виникнення припадків після заплющування очей при дії яскравого світла. Діти часто закривають очі долонями або потирають повіки, щоб позбавитися дискомфорту та неприємних відчуттів, викликаних припадками. Прогноз і відповідь на лікування при цій формі гірший, ніж при класичній і м’якій формах. Близько 75 % пацієнтів мають середні або тяжкі проблеми з навчанням. Магнітно–резонансна томографія (МРТ) головного мозку, як правило, не виявляє патологічних змін. Часто буває спонтанне покращення перебігу у препубертатному віці;
б) атипова форма — характерними є часті генералізовані тоніко–клонічні напади на першому році життя. Міоклонії очей та інші міоклонії, як правило, проявляються протягом першого року життя. На ЕЕГ реєструється коротка та нерегулярна генералізована спайк–хвильова та поліспайк–хвильова активність частотою 2–3 Гц, що виникає після заплющування очей та супроводжується міоклоніями повік та іноді міоклоніями верхніх кінцівок. Зазвичай є виражена фотосенситивність. Також можуть бути фокальні спалахи спайків або комплексів «спайк — повільна хвиля». МРТ головного мозку, як правило, не виявляє патологічних змін. У дітей часто відмічається затримка когнітивного розвитку на рівні інтелектуального дефіциту середнього ступеня. Антиконвульсанти, що блокують натрієві канали (карбамазепін, ламотриджин, фенітоїн), при цій формі можуть провокувати виникнення абсансів і міоклоній.
2. М’яка форма СД.
При цій формі міоклонії повік, що виникають після заплющування очей, можуть тривати у пацієнта роками, не викликаючи дискомфорту до появи тоніко–клонічних судом або випадкової діагностики стану фахівцем. На ЕЕГ відмічаються епілептиформні зміни, що виникають при закриванні очей та фотостимуляції. Протягом життя може виникати лише кілька генералізованих припадків, у зв’язку з чим ці пацієнти часто відмовляються від лікування. Також у таких пацієнтів може розвиватися звичка кліпати очами без наявності супутніх епілептиформних змін на ЕЕГ.
3. Класична форма СД.
При класичній формі відмічаються виражені міо–клонії повік з девіацією очних яблук угору та ретропульсивними рухами голови, що виникають відразу після заплющування очей, а також поліспайк–хвильова генералізована активність на ЕЕГ. В усіх випадках міоклонії очей поєднуються з абсансами та посилюються при фотостимуляції. У деяких випадках присутній феномен руху голови у напрямку джерела світла. У багатьох пацієнтів є приємні відчуття під час припадків. При відсутності лікування у пацієнтів може розвиватися автопровокація припадків шляхом заплющування очей під час періодів зниженої активності або нудьги.
У вітчизняній науковій літературі опис клінічної картини та типових електроенцефалографічних змін при синдромі Дживонса наводять С.К. Євтушенко зі співавторами [15].
Етіологія
Причина виникнення СД на сьогодні лишається остаточно невідомою, проте, найбільш імовірно, захворювання є генетично детермінованим. У більшості зареєстрованих пацієнтів присутній сімейний анамнез захворювання. Також відомі сімейні випадки та випадки захворювання у монозиготних близнюків [16]. У дослідженні закордонних авторів, 28 % пацієнтів мали сімейний анамнез епілепсій, найчастіше синдрому Дживонса, або інших форм ідіопатичних генералізованих епілепсій, а 9,5 % мали фебрильні судоми [17]. Найбільш імовірно, у патогенезі СД відіграє значну роль залучення зорової кори лобних ділянок, потиличної кори та таламуса. При довільному заплющуванні очей, яке регулюється лобними ділянками, або фотостимуляції виникає збудження і син–хронізація нейронів потиличної кори та поширення імпульсу по підкіркових трактах (у тому числі ретикулоталамічних шляхах) на таламус, який генерує та поширює збудження по корі у вигляді пік–хвильової активності [1, 5].
Діагностика
На думку деяких авторів [14], СД відноситься до захворювань, які достатньо один раз побачити протягом життя, і у подальшій практиці діагноз може бути не таким складним, оскільки типові зміни на ЕЕГ та фотосенситивність є патогномонічними для цього синдрому. Наявність міоклоній повік є типовим для СД видом припадків і вагомим приводом, для того щоб запідо–зрити цей діагноз. Panayiotopoulos виділяє таку тріаду симптомів СД [8, 12]:
1) міоклонії повік з наявністю або відсутністю абсансів;
2) провокація припадків і пароксизмальних змін на ЕЕГ заплющуванням очей (fixation–off sensetivity);
3) фотосенситивність.
Діагноз СД базується на даних анамнезу, клінічного спостереження, наявності провокації припадків заплющуванням очей (феномени eyes closure sensitivity та fixation–off sensitivity) та даних ЕЕГ. Для підтвердження діагнозу необхідно проведення відеоелектроенцефалографії (відео–ЕЕГ) — єдиного методу, що дозволяє виявити генералізовану епілептичну активність, яка виникає негайно після заплющування очей. Відео–ЕЕГ виявляє часті та короткі (2–3 секунди) високоамплітудні генералізовані комплекси «спайк — хвиля» з частотою 3–6 Гц або частіше поліспайк–хвильову активність після заплющування очей, що є патогномонічним симптомом захворювання, але можуть виникати також при розплющених очах під дією періодичної ритмічної фотостимуляції або гіпервентиляції. Звертаємо увагу на те, що епілептична активність не виникає, якщо заплющування очей відбувається у темному приміщенні. Також можлива реєстрація епілептиформних змін під час сну без клінічних феноменів. Фотопароксизмальні відповіді реєструються майже в усіх пацієнтів молодого віку, які не отримують терапію. Корисним є проведення відео–ЕЕГ під час сну та пробудження після депривації сну, а також функціональних проб у вигляді гіпервентиляції та періодичної ритмічної фотостимуляції [18]. При СД реєструються епілептифомні зміни, що виникають після заплющування очей, а також провокація припадків та змін на ЕЕГ при сонливості, гіпервентиляції та фотостимуляції [13, 14].
Під час сну зазвичай відбувається значне зниження індексу генералізованої поліпік–хвильової активності або повна її редукція, однак у деяких випадках може зберігатися пік–хвильова активність або фокальні спайки чи комплекси «спайк — хвиля». Максимальний індекс генералізованої поліпік–хвильової активності реєструється після ранкового насильницького пробудження, коли у більшості пацієнтів спостерігаються тривалі групи розрядів дифузних білатерально–асинхронних комплексів поліспайк, комплексів «спайк — хвиля», «поліспайк — хвиля» у поєднанні з дифузними повільними хвилями [13, 14].
Таким чином, діагноз встановлюється у пацієнтів з частими міоклоніями повік та наявністю або відсутністю абсансів, за наявності фотосенситивності та генералізованої активності на ЕЕГ, яка виникає після заплющування очей. При цьому напади мають починатися у дитинстві [1].
Диференційна діагностика
Синдром Дживонса необхідно диференціювати з іншими формами ідіопатичних, криптогенних і симптоматичних епілепсій, що супроводжуються міоклоніями повік або провокацією припадків заплющуванням очей. Серед них ювенільна абсансна епілепсія, ювенільна міоклонічна епілепсія (ЮМЕ) та ідіопатична фотосенситивна потилична епілепсія. Також необхідна диференціація з епілептичними синдромами, що супроводжуються міоклоніями та абсансами: епілепсією з міоклонічними абсансами (синдром Тассинарі) та синдромом періоральних міоклоній з абсансами. Також необхідно відрізняти епілептичні міоклонії повік від пароксизмів неепілептичного походження, зокрема –моторних тиків. Часто пацієнти з міоклоніями повік можуть робити різноманітні гримаси з метою уникнення заплющування очей, які викликають напади. Такі гримаси зазвичай розцінюються як моторні тики або манеризми, що призводять до помилкового направлення пацієнта до психіатра.
Складною є диференційна діагностика між СД та ювенільною міоклонічною епілепсією, при якій також можуть відмічатися міоклонії повік та інших груп м’язів у поєднанні з генералізованою епілептиформною активністю, а також генералізованими тоніко–клонічними припадками. Відмінність полягає у появі при синдромі Дживонса епілептиформної активності, яка виникає негайно після заплющування очей, синхронно з міоклоніями повік, особливо під час ритмічної фотостимуляції [5, 13, 14]. Деякі дослідники вважають СД та ЮМЕ спорідненими синдромами, які можуть трансформуватися один в одний [19]. Описані також випадки трансформації роландичної епілепсії у синдром Дживонса у пубертатному періоді [20].
Міоклонії повік також можуть спостерігатися при інших формах ідіопатичних генералізованих епілепсій з абсансами, симптоматичній абсансній епілепсії, доброякісній міоклонічній епілепсії немовлят. При дитячій абсансній епілепсії абсанси не провокуються заплющуванням очей, тривають довше ніж 3 секунди, можуть супроводжуватися відведенням очей вбік, нехарактерною є також поява епілептиформної активності після заплющування очей та її зникнення після розплющування очей. При інших формах міоклонічних епілепсій можуть відмічатися міоклонії повік без підведення очей угору. Також при ідіопатичній потиличній епілепсії можливе виникнення міоклоній повік під час вторинно–генералізованих припадків.
Міоклонії повік також можливі при криптогенних і симптоматичних епілепсіях, однак при СД зазвичай відсутня затримка розвитку, неврологічна симптоматика та зміни на МРТ, а також уповільнення основного ритму ЕЕГ.
Лікування
На сьогодні в літературі відсутні дані щодо антиконвульсивного препарату, який міг би бути єдиним у лікуванні СД. Ще у 80–х роках минулого сторіччя Сovanis et al. [21] вважали найбільш ефективними вальпроати. З часом у рекомендаціях різних авторів були бензодіазепіни (клоназепам), етосукцимід, ламотриджин і зонізамід. Ті ж Сovanis et al. [22] у 2000–х роках вказували на ефективність леветирацетаму. Хоча леветирацетам має антиміоклонічну й антифотосенситивну дію, контроль припадків при терапії леветирацетамом відмічається лише у 17 % [23]. Однак леветирацетам є кращою альтернативою пацієнтам, особливо для дівчат, враховуючі здатність викликати полікістоз яєчників та тератогенний ефект вальпроатів під час вагітності.
Генералізовані напади при синдромі Дживонса переважно добре відповідають на антиконвульсивну терапію, однак міоклонус повік може залишатися резистентним до лікування. Тому у деяких випадках необхідна політерапія антиконвульсантами. Нерідкими є також випадки фармакорезистентності. Чим раніше та адекватніше розпочато лікування, тим краще відповідь на терапію та менші когнітивні порушення. Деякі фахівці рекомендують комбінацію вальпроатів та етосукциміду, а також додавання невеликих доз ламотриджину до вальпроатів. Монотерапія ламотриджином може провокувати міоклонії. Також описані випадки позитивного впливу клоназепаму у поєднанні з невеликими дозами ламотриджину [25]. Протипоказані карбамазепін, окскарбамазепін, вігабатрін, тіагабін, прегабалін та фенітоїн. Для уникнення фотосенситивності пацієнтам виписують окуляри з блакитним склом, які зменшують частоту нападів.
Необхідно уникати провокуючих факторів, таких як депривація сну, вживання алкоголю, перевтома, тривале використання гаджетів та перегляд телевізору.
Прогноз
Незважаючи на позитивний загальний прогноз, часто синдром Дживонса є пожиттєвим захворюванням, що потребує постійної терапії. Найчастіше у дорослих залишаються резистентні до терапії міоклонії повік без абсансів та фотосенситивності.
Наводимо клінічне спостереження пацієнта з синдромом Дживонса.
Хлопчик, 17 років, звернувся до відділення дитячої психоневрології ДУ «Інститут педіатрії, акушерства і гінекології ім. акад. О.М. Лук’янової НАМН» зі скаргами батьків на наявність у хлопця пароксизмів кліпання та посмикування повік з частотою 3–4 епізоди на 1 годину (до 100 разів на добу), які супроводжуються посмикуванням м’язів шиї та верхніх кінцівок. Також оточуючі помічають у хлопця епізоди «застигання» та «відключення», під час яких відбувається зупинка мовлення. Такі напади пацієнт не пам’ятає, однак після них забуває свою поточну думку.
Перинатальний анамнез пацієнта був не ускладненим. Розвивався відповідно до віку. У школі до останнього часу встигав добре, регулярно займається спортом. У 14 років вперше батьками відмічені епізоди «відключення». У віці 15 років було діагностовано епілепсію з абсансами та призначено препарат вальпроату натрію, який пацієнт приймав протягом року. Через рік до терапії додано бензонал.
За даними огляду вогнищева неврологічна симптоматика відсутня. Присутні скарги на зниження уваги та пам’яті, зниження успішності у навчанні протягом останніх місяців. МРТ головного мозку — без вогнищевих змін.
Для уточнення форми епілепсії хлопчик був направлений на відео–ЕЕГ–моніторинг, наводимо його результати.
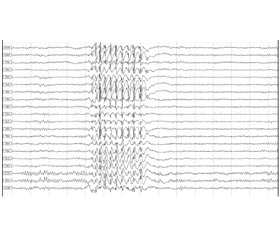
Фонова активність в межах вікової норми. Епілептиформна активність проявляється при заплющених очах у вигляді генералізованих розрядів комплексів «спайк — поліспайк — хвиля» частотою 2,6–3,5 Гц, амплітудою до 1100 мкВ з фронтальним переважанням, тривалістю 1–6 секунд. Синхронно з епілептиформною активністю на ЕЕГ відмічається легке кліпання з поступовим розплющуванням очей (рис. 1).
/37-1.jpg)
У момент нападу падіння м’язового тонусу кінцівок не відзначається, звернене мовлення чує, на запитання відповідає. Також відмічається провокація нападів фотостимуляцією і гіпервентиляцією. Тривалість розрядів при гіпервентиляції збільшується до 7 секунд. З розплющеними очима відзначаються розряди тривалістю до 1 секунди без клінічних проявів.
Таким чином, враховуючи клінічні прояви та результати ЕЕГ, а саме наявність у пацієнта міоклоній повік у поєднанні з епілептичною активністю, що виникає при заплющуванні очей, був встановлений діагноз «синдром Дживонса». Пацієнту було призначено леветирацетам у дозі 30 мг/кг двічі на день. У динаміці через рік на фоні терапії леветирацетамом відмічалося суттєве зменшення міоклоній повік, відсутність абсансів.
Висновки
Синдром Дживонса є захворюванням, що не так часто діагностується в Україні. Це пов’язано зі складністю диференційної діагностики, зокрема схожістю симптомів декількох епілептичних синдромів із міоклонічними нападами, а також низькою настороженістю лікарів щодо даного синдрому.
Для виявлення даної форми епілепсії необхідно модифікувати протокол дослідження і проводити фотостимуляцію з розплющеними й заплющеними очима з частотою стимуляції від 1 до 60 Гц, а також пробу з серією розплющення–заплющення очей в затемненому й освітленому приміщенні. Рекомендується проводити ЕЕГ–дослідження тривалістю не менше 30 хвилин.
Що стосується терапевтичної тактики, можемо свідчити, що, за даними літератури і нашими власними спостереженнями, не існує жодного антиконвульсанта, який міг би бути єдиним у лікуванні СД. Зменшення частоти нападів, але відсутність повної ремісії тільки підтверджує погляд на резистентність СД до антиконвульсивної терапії.
Конфлікт інтересів. Автори заявляють про відсутність конфлікту інтересів при підготовці даної статті.
1. Striano S. Eyelid myoclonia with absences (Jeavons syndrome): a well–defined idiopathic generalized epilepsy syndrome or a spectrum of photosensitive conditions? Epilepsia. 2009 May. 50(Suppl. 5). P. 15–9. doi: 10.1111/j.1528–1167.2009.02114.x.
2. http://www.orpha.net/consor/cgi–bin/OC_Exp.php?lng= EN&Expert=139431.
3. Jeavons P.M. Nosological problems of myoclonic epilepsies in childhood and adolescence. Dev. Med. Child. Neurol. 1977. 19. P. 3–8.
4. Radovici M.M.A., Misirliou V.L., Gluckman M. Epilepsy reflex provoquee par excitations optiques des rayons solaires. Revue Neurologique. 1932. 1. P. 1305–1307. [Translated by M. Koutrou–manidis. Reflex epilepsy provoked by optic excitation by (means of) sunrays. J.S. Duncan, C.P. Panayiotopoulo. Eyelid myoclonia with absences. John Libbey, London, 1996. P. 103–105.]
5. Covanis A. Jeavons syndrome — updated review. Journal of Epileptology. 2015. Vol. 23, № 2. Р. 113–123. https://www.degruyter.com/downloadpdf/j/joepi.2015.23.issue–2/joepi–2015–0033/joepi–2015–0033.pdf.
6. Striano S., Striano P., Nocerino C., Boccella P., Bilo L., Meo R. et al. Eyelid myoclonia with absences: an overlooked epileptic syndrome. Neurophysiol. Clin. 2002. 32. Р. 287–296.
7. Duncan J.S., Panayiotopoulos C.P. Eyelid myoclonia with absences. London: John Libbey and Company Ltd, 1996.
8. Panayiotopoulos C.P., Engel J. Eyelid myoclonia with and without absences. Originally released. July 13, 1999; last updated April 18, 2017; expires April 18, 2020.
9. Commission on Classification and Terminology of the International League Against Epilepsy, 2014.
10. Fisher R.S. et al. Operational classification of seizure types by the International League Against Epilepsy: Position Paper of the ILAE Commission for сlassification and Terminology. Epilepsia. 2017. 58(4). Р. 522–530. doi: 10.1111/epi.13670.
11. Scheffer І.E. et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017. 58(4). Р. 512–521. doi: 10.1111/epi.13709.
12. Panayiotopoulos C.P. Reflex seizures and reflex epilepsies. The epilepsy seizures, syndromes and management. Bladon Medical Publishing, Oxford, 2005. Р. 449–496.
13. Covanis A. Eyelid myoclonia and absences / A.V. Delgado–Escuata, R. Guerrini, M.T. Medina, P. Genton, M. Bureau, C. Dravet. Myoclonic epilepsies, Advances in neurology. 2005. 95. Р. 183–194.
14. Covanis A. Jeavons syndrome. C.P. Panayiotopoulos. Atlas of Epilepsies. Springer–Verlag, London Ltd, 2010. Р. 1081–1091.
15. Евтушенко С.К. Клиническая электроэнцефалография у детей [Текст]: руководство для врачей. Донецк: Донеччина, 2005. 859 с.
16. Yang T., Liu Y., Liu L., Yan B., Xhang Q., Zhou D. Absences status epilepticus in monozygotic twins with Jeavons syndrome. Epileptic. Disord. 2008. 10. Р. 227–230.
17. Caraballo R.H., Fontana E., Darra F., Chacon S., Ross N., Fiorini E., Fejerman N. et al. A study of 63 cases with eyelid myoclonia with or without absences: Type of seizure or an epileptic syndrome? Seizure. 2009. 18. Р. 440–445.
18. Kasteleijn–Nolst Trenite D., Rubboli G., Hirsch E., Martins da Silva A., Seri S., Wilkins A. et al. Methodology of photic stimulation revisited: Updated European algorithm for visual stimulation in the EEG laboratory. Epilepsia. 2012. 53. Р. 16–24.
19. Destina YalÅin A., Forta H., KiliÅ E. Overlap cases of eyelid myoclonia with absences and juvenile myoclonic epilepsy. Seizure. 2006. 15. Р. 359–365.
20. Миронов М.Б., Мухин К.Ю., Петрухин А.С. Трансформация роландической эпилепсии в синдром Дживонса (два клинических случая). Русский журнал детской неврологии. 2009. 4(4). Р. 14–21.
21. Covanis A., Jeavons P.M., Gupta A.K. Sodium valproate: monotherapy and polytherapy. Epilepsia. 1982. 23. Р. 693–720.
22. Covanis A., Katsalouli M. Levetiracetam Monotherapy in Generalized Epilepsy and Photosensitivity in Children and Young Adults. Epilepsia. 2004. 45(Suppl.). 139 р.
23. Кулагин В.Н. К вопросу диагностики и лечения синдрома Дживонса. Русский журнал детской неврологии. 2014. № 2. С. 55–59.