
Журнал «Медицина неотложных состояний» Том 16, №1, 2020
Вернуться к номеру
Расслаивающая аневризма аорты как одна из ведущих причин летальности при синдроме Марфана на примере клинического случая
Авторы: Шмидт Е.Ю.(1), Прилуцкая Е.Ю.(1, 2), Грицай А.О.(1, 2)
(1) — Харьковский национальный университет им. В.Н. Каразина, г. Харьков, Украина
(2) — Харьковская клиническая больница на железнодорожном транспорте № 1, филиал «Центр охраны здоровья» акционерного общества «Украинская железная дорога», г. Харьков, Украина
Рубрики: Медицина неотложных состояний
Разделы: Справочник специалиста
Версия для печати
Синдром Марфана поряд з іншими спадковими захворюваннями сполучної тканини посідає друге місце серед причин аневризми аорти. Прогноз пацієнтів з аневризмою аорти залежить від ранньої діагностики та, відповідно, раннього хірургічного лікування. У нашій статті подано клінічний випадок пацієнтки з пізно діагностованим синдромом Марфана, що був ускладнений на момент діагностики розшаруванням аневризми аорти і в подальшому (на тлі нестабільних цифр артеріального тиску) — розривом висхідного відділу аорти.
Синдром Марфана наряду с другими наследственными заболеваниями соединительной ткани занимает второе место среди причин аневризмы аорты. Прогноз пациентов с аневризмой аорты зависит от ранней диагностики и, соответственно, раннего хирургического лечения. В нашей статье представлен клинический случай пациентки с поздно диагностированным синдромом Марфана, осложнившимся к моменту диагностики расслоением аневризмы аорты и впоследствии (на фоне нестабильных цифр артериального давления) — разрывом восходящего отдела аорты.
Marfan syndrome, along with other hereditary connective tissue diseases, is the second most common cause of aortic aneurysm. The prognosis of patients with aortic aneurysm depends on early diagnosis and, consequently, early surgical treatment. This article presents a clinical case of late diagnosed Marfan syndrome that was complicated by aortic dissection and subsequently (on the background of unstable blood pressure) — by rupture of the ascending aorta.
синдром Марфана; спадкові захворювання сполучної тканини; аневризма аорти; розшарування аорти; розрив аневризми аорти
синдром Марфана; наследственные заболевания соединительной ткани; аневризма аорты; расслоение аорты; разрыв аневризмы аорты
Marfan syndrome; hereditary connective tissue diseases; aortic aneurysm; aortic dissection; ruptured aneurysm
Введение
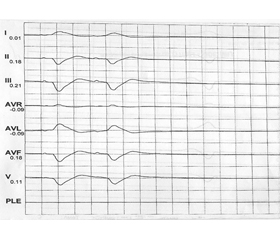
Клинический случай
/125.jpg)
/126.jpg)
/126_2.jpg)
Выводы
1. Фищенко Я.В. Синдром Марфана (обзор литературы). Вісник ортопедії, травматології та протезування. 2013. № 1. С. 66-72. URL: http://nbuv.gov.ua/UJRN/Votip_2013_1_18 (дата звернення: 14.11.2019).
2. Жураєв Р.К. Синдром Марфана: еволюція діагностичних критеріїв. Український медичний часопис. 2012. № 1. С. 98-102. URL: http://nbuv.gov.ua/UJRN/UMCh_2012_1_27 (дата звернення: 14.11.2019).
3. Erbel R., Aboyans V., Boileau C. et al.; ESC Committee for Practice Guidelines. 2014 ESC Guidelines on the diagnosis and treatment of aortic diseases: Document covering acute and chronic aortic diseases of the thoracic and abdominal aorta of the adult. The Task Force for the Diagnosis and Treatment of Aortic Diseases of the European Society of Cardiology (ESC). Eur. Heart J. 2014. Vol. 35. № 41. P. 2873-2926. DOI: http://dx.doi.org/10.1093/eurheartj/ehu281. PubMed PMID: 25173340.
4. Pearson G.D., Devereux R., Loeys B. et al.; National Heart, Lung, and Blood Institute and National Marfan Foundation Working Group. Report of the National Heart, Lung, and Blood Institute and National Marfan Foundation Working Group on research in Marfan syndrome and related disorders. Circulation. 2008. Vol. 118. № 7. P. 785-791. DOI: http://dx.doi.org/10.1161/CIRCULATIONAHA.108.783753. PubMed PMID: 18695204; PubMed Central PMCID: PMC2909440.
5. Baumgartner H., Bonhoeffer P., De Groot N.M. et al.; Task Force on the Management of Grown-up Congenital Heart Disease of the European Society of Cardiology (ESC); Association for European Paediatric Cardiology (AEPC); ESC Committee for Practice Guidelines (CPG). ESC Guidelines for the management of grown-up congenital heart disease (new version 2010). Eur. Heart J. 2010. Vol. 31. № 23. P. 2915-2957. DOI: http://dx.doi.org/10.1093/eurheartj/ehq249. PubMed PMID: 20801927.
6. You W., Hong Y., He H. et al. TGF-β mediates aortic smooth muscle cell senescence in Marfan syndrome. Aging (Albany NY). 2019. Vol. 11. № 11. P. 3574-3584. DOI: https://doi.org/10.18632/aging.101998. PubMed PMID: 31147528; PubMed Central PMCID: PMC6594817.
7. Клеменов А.В., Суслов А.С. Наследственные нарушения соединительной ткани: современный подход к классификации и диагностике (обзор). Современные технологии в медицине. 2014. Т. 6. № 2. С. 127-137. URL: http://www.stm-journal.ru/ru/numbers/2014/2/1065 (дата обращения: 12.11.19).
8. Groenink M., Lohuis T.A., Tijssen J.G. et al. Survival and complication free survival in Marfan's syndrome: implications of current guidelines. Heart. 1999. Vol. 82. № 4. P. 499-504. PubMed PMID: 10490568; PubMed Central PMCID: PMC1760285.
9. Кузьмичев Д.Е., Чирков С.В., Ильина М.П., Вильцев И.М. К вопросу диагностики аневризмы грудной части аорты. Проблемы экспертизы в медицине. 2013. № 3(51). С. 40-43. URL: https://cyberleninka.ru/article/n/k-voprosu-diagnostiki-anevrizmy-grudnoy-chasti-aorty/viewer (дата обращения: 12.11.2019).
10. Грознова О.С., Калачанова Е.П., Леонтьева И.В., Ржевская П.Е. Тяжелое поражение сердечно-сосудистой системы у больного с синдромом Марфана. Российский вестник перинатологии и педиатрии. 2015. № 60(2). С. 95-99. DOI: http://dx.doi.org/10.21508/1027-4065-2019-64-1-116-119.
11. Von Kodolitsch Y., Rybczynski M., Vogler M. et al. The role of the multidisciplinary health care team in the management of patients with Marfan syndrome. J. Multidiscip. Healthc. 2016. Vol. 9. P. 587-614. Review. PubMed PMID: 27843325; PubMed Central PMCID: PMC5098778.
12. Pearson G.D., Devereux R., Loeys B. et al.; National Heart, Lung, and Blood Institute and National Marfan Foundation Working Group. Report of the National Heart, Lung, and Blood Institute and National Marfan Foundation Working Group on research in Marfan syndrome and related disorders. Circulation. 2008. Vol. 118. № 7. P. 785-791. DOI: http://dx.doi.org/10.1161/CIRCULATIONAHA.108.783753. PubMed PMID: 18695204; PubMed Central PMCID: PMC2909440.
13. Hiratzka L.F., Bakris G.L., Beckman J.A. et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with Thoracic Aortic Disease: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine. Circulation. 2010. Vol. 121. № 13. P. e266-369. DOI: http://dx.doi.org/10.1161/CIR.0b013e3181d4739e. PubMed PMID: 20233780.
14. Aubart M., Gazal S., Arnaud P. et al. Association of modifiers and other genetic factors explain Marfan syndrome clinical variability. Eur. J. Hum. Genet. 2018. Vol. 26. № 12. P. 1759-1772. DOI: 10.1038/s41431-018-0164-9. PubMed PMID: 30087447; PubMed Central PMCID: PMC6244213.
15. Марковский В.Д., Сорокина И.В., Калужина О.В. и др. Расслаивающая аневризма аорты у пациента с синдромом Марфана (наблюдение из практики врача-патологоанатома). Актуальні проблеми сучасної медицини: Вісник української медичної стоматологічної академії. 2013. Т. 13. № 4(44). С. 206-210. URL: http://nbuv.gov.ua/UJRN/apsm_2013_13_4_53 (дата обращения: 19.11.2019).
16. Von Kodolitsch Y., De Backer J., Schüle H. et al. Perspectives on the revised Ghent criteria for the diagnosis of Marfan syndrome. Appl. Clin. Genet. 2015. Vol. 8. P. 137-155. DOI: http://dx.doi.org/10.2147/TACG.S60472. Review. PubMed PMID: 26124674; PubMed Central PMCID: PMC4476478.
17. Магомедов М.П., Гаджиев А.Н., Шихабидов Н.Р. и др. Расслоение аорты, осложненное спинальным ишемическим инсультом. Медицинский вестник МВД. 2016. Т. 82. № 3. С. 25-28. URL: http://www.ormvd.ru/pubs/105/came-out-no-3-2016-volume-lxxxii-the-scientific-practical-journal-medical-bulletin-of-the-ministry-o/ (дата обращения: 14.11.2019).
18. Сухарева Г.Э. Сердечно-сосудистые осложнения синдрома Марфана у детей Крымского региона. Российский вестник перинатологии и педиатрии. 2019. Т. 64. № 1. С. 116-119. DOI: http://dx.doi.org/10.21508/1027-4065-2019-64-1-116-119.
19. Тимофеев Е.В., Малев Э.Г., Лунева Е.Б., Земцовский Э.В. Активность трансформирующего фактора роста β у лиц молодого возраста с марфаноидной внешностью. Педиатр (Санкт-Петербург). 2019. Т. 10. № 1. С. 49-56. DOI: https://doi.org/10.17816/PED10149-56.
20. De Beaufort H.W.L., Trimarchi S., Korach A. et al. Aortic dissection in patients with Marfan syndrome based on the IRAD data. Ann. Cardiothorac. Surg. 2017. Vol. 6. № 6. P. 633-641. DOI: http://dx.doi.org/10.21037/acs.2017.10.03. PubMed PMID: 29270375; PubMed Central PMCID: PMC5721116.