Вступ
У практиці лікаря-інтерніста одним із найскладніших досі залишається діагноз амілоїдозу. Найбільш часта локалізація амілоїдозу — нирки, де він виявляється, за даними А.А. Демина (1970), в 1,4 % випадків, за даними секційних спостережень 60–70-х років Г.П. Шульцева (1970) — в 1,9 % випадків [22]. Лікарю-нефрологу швидко встановити цей діагноз не вдається, оскільки доводиться диференціювати з хронічним гломерулонефритом, діабетичною нефропатією, іншими захворюваннями, що супроводжуються розвитком нефротичного синдрому (системні захворювання сполучної тканини тощо) [1]. Провідним диференціально-діагностичним критерієм є морфологічне дослідження тканини, отриманої шляхом біопсії нирки, яка не є рутинною процедурою, що, на жаль, гальмує час до встановлення остаточного діагнозу та призначення відповідного лікування.
За визначенням, амілоїдоз (код МКХ-10: E85) — це поєднана група захворювань, які характеризуються позаклітинним відкладенням специфічного нерозчинного фібрилярного білка амілоїду. Поширеність амілоїдозу достеменно невідома: так, у США частота амілоїдозу становить від 5,1 до 12,8 випадку на 100 000 населення на рік [2]. У країнах третього світу, за думкою S.Y. Tan (1995), смертність від AL-амілоїдозу становить 1 на 2000 населення (0,05 %). У Європі АА-амілоїдоз розвивається у 5 % хворих на хронічні запальні захворювання (частота його в Іспанії — 1,9 % автопсій, у Португалії — 1,4 %,); в Ізраїлі — в 0,55 %, в Японії — тільки в 0,1 %. За іншими джерелами, амілоїдоз ускладнює перебіг ревматоїдного артриту в 6–10 % випадків. У середньому частка амілоїдної нефропатії в структурі захворювань нирок становить 2,5–2,8 %, а в структурі хвороб, що призводять до хронічної ниркової недостатності (ХНН), 1 % (за даними Європейської асоціації діалізу і трансплантації) [3].
Уперше ця патологія була описана в XVII ст. Боне: «сагова» селезінка у хворого на абсцес печінки. Сам термін «амілоїд» був запропонований німецьким патологоанатомом Р. Вірховим у 1853 р., який досліджував речовину, що відкладається в органах хворих на туберкульоз, сифіліс, лепру тощо, і поєднував ці випадки у так звану «сальну хворобу» через те, що знаходив у різноманітних органах речовину, схожу на крохмаль, завдяки її характерній реакції із йодом. У двадцяті роки XX століття Бенхольд запропонував забарвлення амілоїду конго червоним, потім був виявлений ефект подвійного променезаломлення в поляризованому світлі: зміна цегляно-червоного забарвлення на яблучно-зелене в поляризованому світлі.
У 1959 р. Коген і Калкінс за допомогою електронної мікроскопії встановили фібрилярну структуру амілоїду. Гістохімічне дослідження показало, що амілоїд — це складна речовина білкової природи, глікопротеїд, у якому білки щільно зв’язані із полісахаридами, які становлять лише до 4 % маси. Однак історичний термін «амілоїд» залишився у медичній класифікації хвороб [4–6].
У подальшому зміни уявлень про амілоїдоз були засновані на уточненні природи амілоїдних фібрил — особливих білкових структур діаметром 5–10 нм і довжиною до 800 нм, що складаються з 2 і більше паралельно розташованих філаментів. Для білкових субодиниць амілоїдних фібрил характерна своєрідна просторова орієнтація молекули — крос-β-складчаста конформація. З β-складчастою конфігурацією фібрили пов’язана стійкість амілоїду до протеолітичних ферментів міжклітинного матриксу, що обумовлює його значне накопичення з прогресуючим руйнуванням ураженого органа і втратою його функції. Незважаючи на неоднорідність амілоїдних фібрил (глікопротеїни), серед амілоїдогенних факторів провідну роль відводять конформаційній лабільності білків-попередників амілоїду, специфічних для кожного типу амілоїдозу, вміст яких у фібрилі досягає 80 %. Серед інших білків амілоїду особливе значення має так званий амілоїдний P-компонент — похідне білка гострої фази, синтезованого печінкою, структурно схожого з С-реактивним білком. Здатністю пригнічувати клітинну адгезію пояснюється участь амілоїдного Р-білка в обмеженні запальної реакції і блокаді автоімунітету. У складі амілоїду Р-компонент захищає фібрили від ферментативного руйнування макрофагами-амілоїдокластами.
Незважаючи на відмінність у типах амілоїдного білка, механізми формування амілоїдозу подібні. Основна умова розвитку хвороби — наявність певного амілоїдогенного попередника у підвищеній кількості. Поява або посилення амілоїдогенності можуть бути обумовлені молекулярною гетерогенністю білків-попередників (варіантні транстиретини, легкі ланцюги із заміною амінокислот, різні ізотопи білка SAA) і, як наслідок, циркуляцією варіантів білків із підвищеною загальною гідрофобністю молекули і порушеним співвідношенням поверхневих молекулярних зарядів, що призводить до нестабільності білкової молекули і сприяє її агрегації в амілоїдних фібрилах. На останньому етапі амілоїдогенезу відбувається взаємодія амілоїдного білка з білками плазми крові і глікозаміногліканами тканин. При цьому у відкладення амілоїду включаються сироватковий амілоїдний Р-компонент, гепарансульфати і дерматансульфати інтерстиціального глікокаліксу [6].
Клінічні уявлення про амілоїдоз також зазнали значної еволюції: від зв’язку «сальної хвороби» з туберкульозом, сифілісом, рикетсіозом (Рокитанський, 1842), опису «жирних органів» у хворого, який не мав ніяких супутніх захворювань (Вілкс, 1856), виявлення амілоїдозу у хворих із мієломною хворобою (Аткінсон, 1937), виділення старечих (Сойка, 1876) і спадкових (Андраде, 1952; Сєров В.В., 1972; Heller H., 1964) форм, поділу амілоїдозу на генетичний, первинний і вторинний типи і до класифікації ВООЗ 1993 року, побудованої на специфічності основного фібрилярного білка амілоїду після того, як було доведено, що гетерогенність амілоїду обумовлена різноманіттям сироваткових білків-попередників і є зв’язок клінічних форм захворювання з типом цих білків (табл. 1) [6, 7].
За Міжнародною класифікацією хвороб (МКХ-10) амілоїдоз класифікується так:
E85.0 Спадковий сімейний амілоїдоз без невропатії
E85.1 Невротичний спадковий сімейний амілоїдоз
E85.2 Спадковий сімейний амілоїдоз неуточнений
E85.3 Вторинний системний амілоїдоз
E85.4 Обмежений амілоїдоз
E85.8 Інші форми амілоїдозу
E85.9 Амілоїдоз неуточнений
Клінічна класифікація амілоїдозу містить такі форми:
— первинний амілоїдоз:
- виникає без явної причини;
- асоційований із множинною мієломою;
— вторинний амілоїдоз:
- при хронічних інфекціях;
- при ревматоїдному артриті та інших захворюваннях сполучної тканини;
- при онкологічних захворюваннях;
— сімейний (спадковий) амілоїдоз:
- при періодичній хворобі;
- португальський варіант і інші форми сімейного амілоїдозу;
— старечий амілоїдоз;
— локальний амілоїдоз;
— спадковий амілоїдоз:
– невропатичний:
- з ураженням нижніх кінцівок: португальський, японський, шведський та інші типи;
- з ураженням верхніх кінцівок: типи Швейцарія — Індіана, Німеччина — Меріленд;
– нефропатичний:
- періодична хвороба;
- лихоманка і болі в животі у шведів і сицилійців;
- поєднання висипу, глухоти і ураження нирок;
- ураження нирок у поєднанні з артеріальною гіпертензією;
– кардіоміопатичний:
- датський — прогресуюча серцева недостатність;
- мексикансько-американський — синдром слабкості синусового вузла, зупинка передсердь;
– змішаний:
- фінський — дистрофія рогівки і ураження черепно-мозкових нервів;
- мозкові інсульти.
Клінічно значуще ураження нирок спостерігається в основному при вторинному (реактивному) амілоїдозі і при спадкових формах амілоїдозу, перш за все при амілоїдозі, що виникає при періодичній хворобі. В обох випадках амілоїдоз відноситься до АА-типу.
При первинному генералізованому амілоїдозі, хоча і є випадання амілоїду в ниркову тканину, хворі гинуть від серцевої недостатності або від інших причин, тому що нефротичний синдром або хронічна хвороба нирок (ХХН) здебільшого не встигають розвинутися. Проте поява наростаючої протеїнурії за неясної етіології серцевої недостатності, що перебігає з кардіомегалією, може навести на думку про діагноз первинного амілоїдозу, при цьому у багатьох випадках у сечі виявляється білок Бенс-Джонса. У деяких випадках первинного амілоїдозу швидко розвивається нефротичний синдром.
Як правило, набрякам передує досить тривалий доклінічний період. Тому в перебігу амілоїдозу нирок виділяють кілька стадій:
1. Доклінічна (латентна, безсимптомна) стадія, при якій амілоїд присутній в інтермедіарній зоні і по ходу прямих судин пірамідок розвиваються набряк і вогнища склерозу. Стадія триває 3–5 і більше років. У цей період при реактивному амілоїдозі переважають клінічні прояви основного захворювання (наприклад, гнійного процесу в легенях, туберкульозу, ревматоїдного артриту тощо).
2. Протеїнурична (альбумінурична) стадія: амілоїд з’являється насамперед у мезангії, у петлях капілярів, у пірамідках і кірковій речовині гломерул, у судинах. Розвиваються склероз і атрофія нефронів, гіперемія і лімфостаз. Нирки збільшені та щільні, матово-сіро-рожевого кольору. Протеїнурія на початку виражена помірно, потім стає стійкою (стадія переміжної протеїнурії). Тривалість стадії від 10 до 13 років [8–10].
3. Нефротична (набрякла, набряково-гіпотонічна) стадія — амілоїдно-ліпоїдний нефроз: амілоїд у всіх відділах нефрона, є склероз і амілоїдоз мозкового шару, але корковий шар без виражених склеротичних змін. Тривалість стадії до 6 років. Як в протеїнуричній, так і в нефротичній стадії нирки збільшені, щільні (велика сальна нирка). Клінічно ця стадія проявляється класичним нефротичним синдромом з усіма його ознаками. Одним із клінічних диференціально-діагностичних ознак амілоїдного ураження при нефротичному синдромі є системність ураження — виявлення поряд із протеїнурією і анасаркою збільшених лімфатичних вузлів, печінки і селезінки, а також ознак ураження кишечника.
4. Уремічна (термінальна) стадія — амілоїдна зморщена нирка: зменшена в розмірах, щільна, з рубцями нирка. ХНН мало відрізняється від такої при інших захворюваннях нирок. При амілоїдозі азотемія розвивається на тлі низького артеріального тиску і нефротичного синдрому [12].
Клінічна картина дозволяє запідозрити амілоїдоз, однак прижиттєва діагностика амілоїдозу базується на отриманні за допомогою біопсії і дослідженні гістологічного матеріалу різних органів і тканин із забарвленням конго червоним або тіофлавіном. Так, аспіраційна біопсія дозволяє діагностувати амілоїдоз печінки, за даними різних авторів, від 50 до 95 %, і нирки, що інформативно в 85 % випадків [4, 6, 13]. При первинному генералізованому амілоїдозі, коли відбувається периколагенове випадання амілоїду, більш інформативною може виявитися біопсія ясен чи язика. Препарати забарвлюють конго червоним або тіофлавіном Т або S.
Для типування АА- і AL-амілоїдозу гістологічні зрізи органів інкубують в розчині перманганату калію. У результаті АА-білок втрачає спорідненість до конго червоного, у той час як AL-білок — ні. Крім того, AL-амілоїд піддається денатурації після фіксації формаліном, тоді як АА-протеїн не денатурується і тому виявляється імунопероксидазним методом [9, 10, 15].
При первинному амілоїдозі, доброякісній плазмоклітинній дискразії, спорідненій множинній мієломі аномальні клони плазматичних клітин кісткового мозку продукують амілоїдогенні імуноглобуліни. Деякі амінокислоти у варіабельних ділянках легких ланцюгів цих імуноглобулінів займають незвичайну позицію, що призводить до їх нестабільності і схильності до фібрилогенезу. У хворих із первинним амілоїдозом уміст плазматичних клітин у кістковому мозку підвищений до 5–10 % (у нормі їх менше від 4 %), і вони продукують переважний при імуногістохімічному забарвленні ізотип легких ланцюгів імуноглобулінів [7, 15]. Вільні моноклональні легкі ланцюги лямбда- або (рідше) каппа-ізотипу визначаються в крові і сечі, але вміст їх нижче, ніж при мієломній хворобі [8, 16].
Клінічна картина первинного амілоїдозу різноманітна і визначається переважним залученням до патологічного процесу тих чи інших органів: серця, нирок, нервової системи, шлунково-кишкового тракту, печінки та інших. Першими симптомами є слабкість і втрата ваги, але на цій стадії (до появи органних симптомів) діагноз встановлюється вкрай рідко. Органами-мішенями при AL-амілоїдозі найчастіше стають нирки і серце. Ураження нирок проявляється нефротичним синдромом, персистуючим і при настанні ХНН, гематурія й артеріальна гіпертензія не характерні. При відкладенні амілоїду в міокарді розвиваються різноманітні варіанти порушень ритму, прогресуюча серцева недостатність, чому можуть передувати безсимптомні зміни на ЕКГ у вигляді зниження вольтажу зубців. Ехокардіографічне дослідження виявляє концентричне потовщення стінок лівого і правого шлуночків, зменшення об’єму порожнин серця, зниження фракції викиду, діастолічну дисфункцію міокарда лівого шлуночка [8, 16, 17]. Часто відзначаються симптоми ураження нервової системи: вегетативної у вигляді ортостатичної гіпотензії і периферичної у вигляді розладів чутливості. Диспептичні явища і синдром порушеного всмоктування можуть бути обумовлені як ураженням вегетативної нервової системи, так і амілоїдозом шлунково-кишкового тракту. Дуже характерна гепатомегалія, природу якої слід диференціювати між застійними явищами внаслідок серцевої недостатності і амілоїдозом печінки. Селезінка уражається часто, однак спленомегалія виявляється не завжди і великого клінічного значення не має. Рідше зустрічається ураження судин, симптомами якого є періорбітальна пурпура — «очі єнота» і екхімози. Можуть спостерігатися кровотечі, обумовлені як зміною судинної стінки, так і порушенням системи згортання, у першу чергу дефіцитом X-фактора, який зв’язується з амілоїдом. Дефіцитом факторів згортання прийнято пояснювати і характерний для амілоїдозу тромбоцитоз. Макроглосія, класична ознака первинного амілоїдозу, відзначається у 20 % пацієнтів, інфільтрація м’яких тканин може призводити до атрофії м’язів, шкіри, дистрофії нігтів, алопеції і появи пухлиноподібних утворень — амілоїдом. Амілоїдоз легень часто виявляється лише при автопсії. Однак у деяких випадках задишка, кровохаркання і рідина, яка швидко накопичується в плевральній порожнині, можуть бути обумовлені не тільки застійною серцевою недостатністю і/або нефротичним синдромом, а й відкладенням амілоїду в альвеолах або розвитком легеневих амілоїдом. Рентгенологічно можуть виявлятися сітчасті і нодулярні зміни в легеневій тканині. У 10–20 % хворих може мати місце гіпотиреоз як прояв ураження щитоподібної залози.
Діагноз AL-амілоїдозу, крім зазначених клінічних рис, які можуть бути подібними і при вторинному амілоїдозі, базується на низці лабораторних даних. У 85 % пацієнтів при імуноелектрофорезі білків сироватки крові виявляються моноклональні імуноглобуліни. Ті ж моноклональні імуноглобуліни виявляються в сечі у вигляді білка Бенс-Джонса [7, 8]. Біопсія кісткового мозку дозволяє провести диференціальну діагностику з множинною мієломою, а також виявити помірне підвищення кількості плазматичних клітин і їх моноклональність при імуногістохімічному забарвленні. Однак навіть поєднання характерної клінічної картини з наявністю моноклональних плазмоцитів і білків ще не є достатнім для підтвердження діагнозу первинного амілоїдозу. Вирішальну роль відіграють дані біопсії. Найменш інвазивною є аспірація підшкірної жирової клітковини передньої черевної стінки, що дає 80–90 % позитивних результатів при AL-амілоїдозі. Певне діагностичне значення має біопсія ясен і слизової оболонки прямої кишки, але відсоток позитивних результатів широко варіює залежно від стадії процесу, тому доцільно виконання біопсії одного з уражених органів: нирки, печінки, серця, що дає майже 100 % позитивних результатів при амілоїдозі AL-типу.
При виявленні конгофілії досліджуваного матеріалу необхідно його вивчення в поляризованому світлі, ефект подвійного променезаломлення характерний тільки для амілоїду, інші конгофільні речовини яблучно-зеленого забарвлення не дають. Після цього бажано типування амілоїду. Найбільш точним є імуногістохімічний метод з використанням моноклональних антитіл до білків-попередників амілоїду. Тому для діагностики використовуються методи забарвлення за допомогою розчинів лужного гуанідину або перманганату калію, що дозволяють, хоча і побічно, визначити тип фібрилярних відкладень. Обробка препаратів сумішшю розчинів перманганату калію і сірчаної кислоти перед забарвленням конго червоним дозволяє розмежувати АА від усіх інших типів: тільки АА-амілоїд у цьому випадку втрачає конгофілію. Застосування гуанідину змінює конгофільні властивості амілоїду різних типів залежно від часу інкубації і дозволяє диференціювати AA-, ATTR- і AL-амілоїд. Так, якщо після інкубації в розчині гуанідину протягом однієї хвилини конгофілія зникає, це характерно для AA-амілоїду. Якщо ж конгофілія зникає лише після інкубації з розчином гуанідину протягом двох годин — найбільш імовірний старечий амілоїдоз, якщо не зникає і після двох годин — швидше за все, це спадковий варіант. Амілоїд, що сприймає після обробки гуанідином забарвлення конго червоним фрагментарно, найімовірніше, AL-амілоїд [18].
Прогноз при AL-амілоїдозі гірший, ніж при інших формах захворювання, середня тривалість життя не перевищує двох років, при наявності ураження серця або мультисистемного ураження без лікування хворі гинуть протягом декількох місяців [20]. Найбільш частими причинами смерті є серцева недостатність, ниркова недостатність, сепсис, судинні ускладнення і кахексія. Патогенетична схожість з мієломною хворобою дозволяє розраховувати на гальмування прогресування амілоїдозу при хіміотерапії, що проводиться з метою пригнічення моноклональних плазмоцитів [18, 19, 21, 22].
Лікування амілоїдозу є досить складним і не завжди супроводжується позитивним ефектом [9, 10, 20]. У сучасних рекомендаціях надається кілька схем лікування.
1. Циклічне пероральне застосування мельфолану (0,15–0,25 мг/кг маси тіла на добу) і преднізолону (1,5–2,0 мг/кг на добу) по 4–7 днів кожні 4–6 тижнів протягом року до досягнення курсової дози 600 мг.
2. Пероральне застосування мельфолану в дозі 4 мг на добу протягом 3 тижнів, потім після двотижневої перерви — 2–4 мг/добу 4 дні на тиждень постійно до досягнення курсової дози 600 мг у комбінації з преднізолоном.
3. Внутрішньовенне введення високих доз мельфолану (100–200 мг/м2 поверхні тіла протягом 2 днів) з наступною трансплантацією автологічних стовбурових клітин.
4. Внутрішньовенне введення дексаметазону в дозі 40 мг протягом 4 днів кожні 3 тижні — 8 циклів.
5. Внутрішньовенне введення дексаметазону в дозі 40 мг в 1–4, 9–12 і 17–20-й дні 35-денного циклу — 3–6 циклів із подальшим застосуванням інтерферону альфа в дозі 3–6 млн одиниць 3 рази на тиждень.
Застосування високих доз мельфолану з трансплантацією автологічних стовбурових клітин дозволяє досягти ремісії в більше ніж 50 % випадків, однак використання цього методу обмежене тяжкістю стану, віком хворих, функціональними порушеннями з боку серця і нирок. У багатьох випадках можливою виявляється лише симптоматична підтримуюча терапія, що не дозволяє досягти позитивного клінічного ефекту.
Наводимо клінічний приклад, що характеризує труднощі діагностики і лікування хворого на первинний AL-амілоїдоз [11, 12].
Клінічний приклад
Хворий Б., 49 років, надійшов на обстеження і лікування до міського центру нефрології із скаргами на набряки нижніх кінцівок, попереку та живота, швидку стомлюваність, виражену загальну слабкість, запаморочення, зниження апетиту.
З даних анамнезу з’ясовано, що хворим вважає себе з травня 2018 року, коли вперше з’явилися набряки гомілок. Самостійно звернувся до судинного хірурга у червні 2018 р., оглянутий із висновком: вторинна лімфедема гомілок 1-го ступеня, хронічна лімфовенозна недостатність 1-ї ст. Призначений лімфоміозот (Lymphomyosot), серрата (Serratiopeptidase), робив еластичне бинтування нижніх кінцівок, але ефекту не було. У квітні 2018 р. при амбулаторному обстеженні зроблені: еходопплеркардіографія (ЕхоКГ) — без особливостей, фракція викиду ЛШ 67 %; МРТ-обстеження органів черевної порожнини — виявлений жировий гепатоз, дрібні кісти печінки, кіста лівої нирки 5 мм. У серпні 2018 р. оглянутий урологом, але даних за урологічну патологію немає. У той же період у лабораторних аналізах виявлена гіпоальбумінемія до 28 г/л, підвищення загального холестерину до 11,24 ммоль/л, добова протеїнурія становила 3,9 г/л. Дані загального та біохімічного аналізу крові, коагулограми надані у табл. 2, загального аналізу сечі — у табл. 3. Були виключені гепатити В, С, ВІЛ. На рекомендовану консультацію до нефролога не звернувся.
/54.jpg)
Динаміка лабораторного обстеження (загальний аналіз крові, біохімічні показники, коагулограма) хворого наведена у табл.2.
Динаміка загального аналізу сечі, аналізу сечі за Нечипоренком, аналізу сечі за Зимницьким та добового аналізу сечі хворого наведена у табл.3.
У жовтні 2018 р. у хворого сталася шлунково-кишкова кровотеча, з приводу якої лікувався у хірургічному відділенні лікарні швидкої медичної допомоги, де був встановлений діагноз: синдром Мелорі — Вейса. У лабораторних аналізах мали місце лейкоцитоз, гіпопротеїнемія, протеїнурія. При рентген-обстеженні ОГК виявлена позашпитальна пневмонія праворуч, отримував антибактеріальну терапію з позитивною динамікою.
Після виписки з хірургічного стаціонару набряки посилилися до ступеня анасарки. У листопаді 2018 р. був направлений до терапевтичного відділення міської лікарні № 3, де перебував на обстеженні і лікуванні 4 дні. В аналізах крові виявлений лейкоцитоз 12,8 × 109, ШОЕ 36 мм/год, підвищення креатиніну крові, порушення ліпідного обміну, сечовий синдром у вигляді еритроцитурії більше ніж 100 у полі зору. Хворий консультований лікарем-нефрологом і переведений для подальшого обстеження і лікування до міського центру нефрології м. Дніпра. При огляді: зріст 180 см, вага 80 кг. Загальний стан середнього ступеня тяжкості. Шкірні покриви звичайного кольору та вологості. На шкірі тулуба, обличчя множинні пустульозні висипання. На передній поверхні гомілок трофічні порушення шкіри у вигляді пігментації та ділянок депігментації. Видимі слизові блідо-рожеві. Набряки нижніх кінцівок, передньої черевної стінки, попереку. АТ 110/70 мм рт.ст. ЧСС 86 уд/хв. Тони серця приглушені, ритм правильний. При аускультації легень дихання везикулярне, хрипів немає. Язик вологий, обкладений білим нашаруванням. Живіт м’який, безболісний. Печінка збільшена на 2–3 см, край безболісний. Діурез до 1 л/добу.
Хворий був обстежений додатково.
Аналіз крові на ПСА 12.11.2018 р. — 52 %, ПСА total — 0,11 нг/мл; ПСА free — 0,21.
Дослідження маркерів системних захворювань сполучної тканини, системних васкулітів від 08.11.2018 — у нормі.
Імунохімія крові і сечі від 12.11.2018 р.:
— електрофорез сироватки: зниження альбуміну, збільшення альфа- та бета-фракції глобулінів. Нефротичний синдром;
— електрофорез сечі: альбумін, альфа-1-, альфа-2-глобуліни, трансферин та інші сироваткові білки. Неселективна протеїнурія.
Загальний аналіз мокроти від 14.11.2018 р.: слизисто-гнійна, лейк. 20–30 у полі зору, еритроцити 0–1 у полі зору, епітелій бронхів 0–4 у полі зору, альвеолярні клітини 2–5 у полі зору.
Посів мокротиння на мікрофлору та чутливість до антибіотиків від 14.11.18 р.: St. аureus — невелика кількість, Сitrobacter spp., Е. coli — невелика кількість.
ЕКГ від 12.11.2018 р. Ритм синусовий, ЧСС 80–86. Електрична вісь серця відхилена праворуч. Блокада задньої гілки лівої ніжки пучка Гіса.
Рентгенограма органів грудної клітки від 06.11.18 р. У легенях: справа на верхівці щільні вогнища. З обох сторін деформація легеневого рисунка. Корені щільні з кальцинатами, синуси вільні. Діафрагма без особливостей. Серце, аорта — без особливостей.
КТ ОГК від 21.11.2018 р. Висновок: МЗКТ-ознаки остаточних змін після перенесених запалень у легенях, пневмофіброз (наслідки перенесеного туберкульозу).
Консультація фтизіатра від 26.11.2018 р. Даних за активний туберкульозний процес немає. Залишкові зміни після перенесеної пневмонії у нижній частці правої легені. Рекомендоване спостереження у лікаря-пульмонолога.
Консультація дерматолога від 14.11.2018 р. Діагноз: вугровий висип.
Рентгенограма черепа від 06.11.2018 р. У бічній проєкції визначаються в проєкції лобової кістки ділянки просвітлення округлої форми. На прямій рентгенограмі черепа кістково-деструктивних змін не визначається. Звапніння фронтального шва.
Рентгенограма кісток таза: на прямій рентгенограмі кісток таза кістково-травматичних і деструктивних змін не визначається.
УЗД сечового міхура, предміхурової залози від 16.11.2018 р.: слабко виражені дифузні зміни передміхурової залози.
УЗД щитоподібної залози від 16.11.2018 р.: без патологічних змін.
Аналіз крові на гормони щитоподібної залози від 12.11.2018 р.: антитіла до ТПО — 6,4; вільний тироксин — 14,5; ТТГ — 4,3.
Аналіз пунктату кісткового мозку від 20.11.2018 р. У кістковому мозку відзначається наявність 10 % плазматичних клітин.
Хворому був встановлений діагноз: хронічна хвороба нирок 1-ї ст., гломерулонефрит, нефротичний синдром. На підставі додаткового обстеження виключені системні захворювання сполучної тканини, васкуліти, мієломна хвороба, інша онкопатологія. Був запідозрений амілоїдоз, 22.11.2018 р. проведена біопсія підслизового шару ясен. Ознак амілоїдозу в препараті виявлено не було. Для уточнення діагнозу хворий був направлений до ДУ «Інститут нефрології НАМН України» для проведення нефробіопсії, яка була зроблена 04.12.2018 р., встановлений діагноз: ХХН 2-ї ст.: амілоїдоз нирок (АL-амілоїдоз — нефробіопсія 04.12.2018 р.). Нефротичний синдром. Шлунково-кишкова кровотеча (Forest IIв) 13.12.2018 р.. Синдром Мелорі — Вейса.
Подальше обстеження і лікування хворого проводилося в міському центрі нефрології м. Дніпра. При об’єктивному обстеженні у січні 2019 р.: вага 80,2 кг, ІМТ — 24,7 кг/м2. Загальний стан середнього ступеня тяжкості. Шкірні покриви звичайного окрасу та вологості. На шкірі тулуба, обличчя вугровий висип, екхімози. Видимі слизові блідо-рожеві. Набряки нижніх кінцівок, передньої черевної стінки, попереку. АТ 90/60 мм рт.ст. ЧСС 80 на хвилину. Тони серця приглушені, ритм правильний. Дихання везикулярне, хрипів немає. Частота дихання 18 за хвилину. Язик вологий, обкладений білим нашаруванням. Живіт м’який, безболісний. Асциту немає. Печінка виступає з-під краю ребер на 2–3 см, край безболісний.
Хворому проведені в динаміці: ЕхоКГ — суттєвих змін не виявлено, ФВ 65 %, на ЕКГ — ритм синусовий, регулярний, з ЧСС 85 ударів на хвилину. Електрична вісь серця відхилена вправо. Помірно виражені зміни міокарда.
Дані ФЕГДС: вторинна геморагічна гастродуоденопатія. Стан — Форрест 1. Виразкових дефектів не знайдено.
На рентгенограмі ОГК (січень 2019 р.): легеневі поля без вогнищевих і інфільтративних тіней. З обох сторін на верхівці легень вогнищевий пневмофіброз. Корені легень фіброзно ущільнені. Серце та аорта без особливостей.
Хворий оглянутий окулістом, виявлений субкон’юнктивіт. Крововилив ОІ. Ниркова нейроангіопатія.
Проводилося лікування діуретиками, розпочата терапія алкераном 10 мг/добу, медролом 32 мг/добу, хворий отримував альбумін, фраксипарин 0,3 мл підшкірно, інгібітор протонної помпи, розувастатин. Після суттєвого зменшення набряків хворий був виписаний для продовження амбулаторного лікування. Рекомендована консультація нефролога через місяць із аналізами в динаміці.
Надалі хворий 3 місяці лікувався амбулаторно, у травні 2019 р. — у центрі нефрології, але, незважаючи на лікування, зберігався нефротичний синдром, виражена гіпопротеїнемія, стійка гіпотензія. Діурез 700 мл/добу. Отримував свіжозаморожену плазму, алкеран з 17.05 по 21.05.2019 р., медрол 20 мг/добу з 14.05.2019 р., діуретики, гепато- і нефропротектори, бета-блокатор. Ефект від лікування незначний, виписаний з відділення за наполяганням.
У квітні 2019 р. при госпіталізації загальний стан тяжкий. Вага 90 кг, ІМТ — 27,8 кг/м2. Шкірні покриви бліді, екхімози та вугровий висип на обличчі, тулубі. Видимі слизові блідо-рожеві. Виражені набряки нижніх кінцівок, передньої черевної стінки, попереку, верхніх кінцівок. АТ 90/60 мм рт.ст., ЧСС 100 ударів на хвилину. Тони серця глухі, ритм правильний. Дихання везикулярне, значно послаблене в нижніх відділах. ЧД 22 на хвилину. Язик вологий, обкладений білим нашаруванням. Живіт м’який, безболісний. Печінка виступає на 2–3 см, край безболісний. Діурез 600 мл/добу.
На рентгенограмі ОГК у 2 проєкціях виявлені ознаки лівобічного плевриту. На УЗД плевральних порожнин — з обох сторін вільна рідина. Висота шару справа до 106 мм, зліва до 134 мм.
Пацієнту двічі проведено плевральні пункції. Отриманий транссудат, при патогістологічному дослідженні рідини з плевральної порожнини у препаратах — розрізнені клітини мезотелію, лімфоцити.
ЕКГ (квітень 2019 р.): передшлуночкова тахікардія, пароксизм з ЧСС 135 за 1 хв. Електрична вісь серця вертикальна. Виражені дифузні зміни міокарда.
Хворому протягом 3 діб продовжено лікування діуретиками, алкераном 10 мг/добу, медролом
32 мг/добу, інгібітором протонної помпи (пантопразол 40 мг внутрішньовенно), статином, альбуміном. У зв’язку з повторними плевральними пункціями та можливим інфікуванням плеври проведено 5-денний курс антибактеріальної терапії цефтріаксоном 1 г внутрішньовенно 2 рази на добу. Був виписаний за наполяганням без суттєвої позитивної динаміки, на фоні патогенетичної терапії зберігається виражений нефротичний синдром — стійка гіпопротеїнемія, гіпотензія, патогенетична терапія неефективна.
Через 10 днів доставлений до стаціонару у вкрай тяжкому стані, зі скаргами на набряки нижніх та верхніх кінцівок, мошонки, попереку та живота, швидку стомлюваність, головокружіння, відсутність апетиту, зниження діурезу, підвищення температури тіла до 39 °С. Значне погіршення стану протягом 2 діб. При об’єктивному огляді шкірні покриви бліді, екхімози, вугровий висип. Видимі слизові блідо-рожеві. Виражені набряки нижніх кінцівок, передньої черевної стінки, попереку, верхніх кінцівок. АТ 80/60 мм рт.ст. ЧСС 110 уд/хв. Тони серця глухі, ритм правильний. Дихання везикулярне, різко ослаблене в нижніх відділах.
ЧД 24/хв. Язик вологий, обкладений білим нашаруванням. Живіт м’який, чутливий при пальпації. Печінка виступає на 3–4 см, край безболісний. Симптом Пастернацького негативний. Діурез 300–400 мл/добу.
Розпочато симптоматичне лікування, спрямоване на корекцію порушень білкового та водно-електролітного обміну, порушень згортання крові, інфузії розчинів альбуміну та свіжозамороженої плазми. Продовжено лікування медролом 16 мг/добу як у зв’язку з гіпотензією, так і в рамках патогенетичної терапії, але у хворого наростала клініка нефротичного кризу (загальний білок 28,3 г/л, гіпотонія, мігруюча та болісна еритема шкіри верхніх кінцівок, біль у животі). На тлі стабільно тяжкого стану гіпотонія посилилася, хворий впав у сопор і помер через 20 годин після госпіталізації.
Основний клінічний діагноз: хронічна хвороба нирок II стадії (ШКФ 60 мл/хв): амілоїдоз нирок (АL-амілоїдоз — нефробіопсія від 2018 р.). Нефротичний синдром.
Ускладнення: нефротичний криз. Гостра серцево-судина недостатність.
Супутні захворювання: дисметаболічна кардіоміопатія складного генезу зі збереженою ФВ ЛШ. Вторинна геморагічна гастродуоденопатія. Синдром Мелорі — Вейса.
Було проведено патологоанатомічне дослідження, при якому підтверджено ідіопатичний системний амілоїдоз (АL-амілоїдоз — нефробіопсія від 2018 р.) із переважним ураженням нирок, печінки, селезінки. Причиною смерті визнана поліорганна недостатність внаслідок тяжких дистрофічно-атрофічних змін внутрішніх органів аж до виснаження, із переважанням серцевої недостатності внаслідок дисметаболічної кардіоміопатії складного генезу, яка ускладнилася двостороннім гідротораксом (по 2200 мл), гідроперикардом (170 мл), анасаркою.
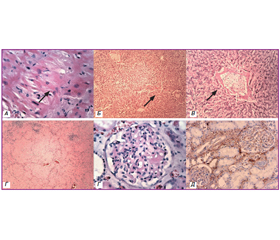
При гістологічному дослідженні в серці спостерігались тяжкі дистрофічно-атрофічні зміни кардіоміоцитів з ознаками втрати міоглобіну, наявність гранул ліпофусцину в цитоплазмі міозитів, однак вірогідних ознак амілоїдозу не виявлено (рис. 1А). Ознаки ліпофусцинозу також були у клітинах печінки. Відкладення амілоїду у вигляді аморфних рожевих мас спостерігались в печінці по портальних трактах та у стінках центральних вен (рис. 1Б, В), масивні відкладення амілоїду були в стромі селезінки, що супроводжувалось вираженою атрофією лімфоїдних фолікулів (рис. 1Г). У нирках накопичення амілоїду спостерігалось по базальних мембранах канальців та судин, капілярів і в мезангії ниркових клубочків, що було підтверджено імуногістохімічно (рис. 1Ґ, Д). Прояви амілоїдозу були також в слизових оболонках шлунково-кишкового тракту і легенях, однак менш виражені.
/56.jpg)
Слід зазначити, що від розвитку системних проявів до смерті хворого минуло близько двох років. Діагноз AL-амілоїдозу був встановлений через пів року від розвитку нефротичного синдрому, але швидкий ступінь прогресування захворювання і зростаюча тяжкість стану хворого не дозволяли проводити високоспецифічне лікування до верифікації діагнозу. Усе це призвело до стрімкого прогресування захворювання із розвитком системних уражень. Найбільш яскраво вираженими були ураження нирок, печінки, шлунково-кишкового тракту і легень (рис. 1). Вірогідних ознак амілоїдного ураження серця не виявлено, мали місце виражені дистрофічно-атрофічні зміни серцевого м’яза.
У клінічній картині на перший план виступали кардіореспіраторні ураження і нефротичний синдром, хоча приєднання ХНН істотно не позначилося на перебігу захворювання. Патогенетична терапія алкераном і медролом була розпочата на тлі розгорнутої клініки системного амілоїдозу у хворого з вираженими білковими і водно-електролітними розладами, коагулопатією і проводилася протягом лише 6 місяців з перервами, курсова доза досягнута не була. Неефективність патогенетичного лікування була обумовлена у першу чергу тяжкістю захворювання і відносно пізнім початком терапії.
Висновки
На підставі наведеного клінічного випадку автори бажали привернути увагу до необхідності своєчасної діагностики та раннього початку лікування цієї складної патології.
Конфлікт інтересів. Автори заявляють про відсутність конфлікту інтересів при підготовці даної статті.

/51.jpg)
/54.jpg)
/56.jpg)