
Международный эндокринологический журнал Том 16, №6, 2020
Вернуться к номеру
Діабетична кардіоміопатія: діагностичні біомаркери
Авторы: Сергієнко В.О., Сергієнко О.О.
Львівський національний медичний університет імені Данила Галицького, м. Львів, Україна
Рубрики: Эндокринология
Разделы: Справочник специалиста
Версия для печати
В огляді наведені класифікація біомаркерів захворювань серцево-судинної системи, біологічні маркери, що знайшли застосування в кардіологічній клініці, біомаркери серцевої недостатності, сучасні рекомендації з використання біомаркерів для діагностики та лікування гострої і хронічної серцевої недостатності. Особливу увагу приділено значенню прогіпертрофічних біомаркерів міокарда (передсердному натрійуретичному пептиду, натрійуретичному пептиду мозку та N-кінцевому фрагменту попередника мозкового натрійуретичного пептиду, кардіотрофіну-1); біомаркерам порушень скоротливої функції міокарда (тропонінам); простеатозним біомаркерам діабетичної кардіоміопатії (серцевому білку, що зв’язує жирні кислоти); значенню епікардіальної жирової тканини; маркерам ремоделювання позаклітинного матриксу (матриксним металопротеїназам); маркерам фіброзу та запальних процесів (трансформуючому фактору росту β, галектину-3, стимулюючому фактору росту ST2). Однак використання біомаркерів для ідентифікації дисфункції лівого шлуночка залишається дискутабельним питанням. Натрійуретичні пептиди вивільняються у відповідь на прогресування стрес-індукованої кардіоміопатії, що рідко спостерігається у пацієнтів із субклінічною дисфункцією та гіпертрофією лівого шлуночка. Ожиріння асоціюється з нижчими рівнями натрійуретичних пептидів, що може погіршити чутливість тесту. Проте скринінг на основі натрійуретичного пептиду є ефективним для виявлення помірної діастолічної дисфункції. Повідомляється також про інші потенційні біомаркери порушень функції міокарда при цукровому діабеті, включаючи циркулюючі мікрорибонуклеїнові кислоти та метаболіти глюкози (наприклад, O-GlcNAc), виявлені в циркулюючих еритроцитах. Однак досі не досягнуто чіткого консенсусу щодо клінічної ролі будь-якого з цих можливих біомаркерів.
В обзоре приведены классификация биомаркеров заболеваний сердечно-сосудистой системы, биологические маркеры, которые нашли применение в кардиологической клинике, биомаркеры сердечной недостаточности, современные рекомендации по использованию биомаркеров для диагностики и лечения острой и хронической сердечной недостаточности. Особое внимание уделено значению прогипертрофических биомаркеров миокарда (предсердного натрийуретического пептида, натрийуретического пептида мозга и N-конечного фрагмента предшественника мозгового натрийуретического пептида, кардиотрофина-1); биомаркеров нарушений сократительной функции миокарда (тропонина); простеатозным биомаркерам диабетической кардиомиопатии (сердечному белку, связывающему жирные кислоты); значению эпикардиальной жировой ткани; маркерам ремоделирования внеклеточного матрикса (матриксным металлопротеиназам); маркерам фиброза и воспалительных процессов (трансформирующему фактору роста β, галектину-3, стимулирующему фактору роста ST2). Однако использование биомаркеров для идентификации дисфункции левого желудочка остается дискутабельным вопросом. Натрийуретические пептиды высвобождаются в ответ на прогрессирование стресс-индуцированной кардиомиопатии, что редко наблюдается у пациентов с субклинической дисфункцией и гипертрофией левого желудочка. Ожирение ассоциируется с более низкими уровнями натрийуретических пептидов и может ухудшить чувствительность теста. Тем не менее скрининг на основе натрийуретического пептида является эффективным для выявления умеренной диастолической дисфункции. Сообщается также о других потенциальных биомаркерах нарушений функции миокарда при сахарном диабете, включая циркулирующие микрорибонуклеиновые кислоты и метаболиты глюкозы (например, O-GlcNAc), обнаруженные в циркулирующих эритроцитах. Однако до сих пор не достигнут четкий консенсус относительно клинической роли этих возможных биомаркеров.
The review provides a classification of biomarkers of the cardiovascular system diseases, biological markers that have found application in a cardiological clinic, biomarkers of heart failure, modern recommendations on the use of biomarkers for the diagnosis and treatment of acute and chronic heart failure. Special attention is paid to the importance of myocardial pro-hypertrophic biomarkers (atrial natriuretic peptide, brain natriuretic peptide, N-amino terminal fragment of the prohormone B-type natriuretic peptide, cardiotrophin-1); biomarkers of myocardial contractile dysfunction (troponins); pro-steatosis biomarkers of diabetic cardiomyopathy (human heart-type fatty acid binding protein); the value of epicardial adipose tissue; extracellular matrix remodeling markers (matrix metalloproteinases); fibrotic and inflammatory biomarkers (transforming growth factor beta, galectin-3, stimulating growth factor ST2). However, the use of biomarkers to identify left ventricular dysfunction remains a debatable issue. Natriuretic peptides are released in response to the progression of stress-induced cardiomyopathy, which is rare in patients with subclinical dysfunction and left ventricular hypertrophy. Obesity is associated with lower levels of natriuretic peptides, which may impair the sensitivity of the test. However, screening based on natriuretic peptide is effective for detecting moderate diastolic dysfunction. Other potential biomarkers of myocardial dysfunction in diabetes have also been reported, including circulating microribonucleic acids and glucose metabolites (eg, O-GlcNAc) found in circulating erythrocytes. However, a clear consensus has not yet been reached on the clinical role of any of these possible biomarkers.
цукровий діабет; діабетична кардіоміопатія; біомаркери; огляд
сахарный диабет; диабетическая кардиомиопатия; биомаркеры; обзор
diabetes mellitus; diabetic cardiomyopathy; biomarkers; review
/13.jpg)
/14.jpg)
/15.jpg)
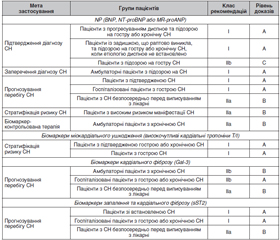
Прогіпертрофічні біомаркери міокарда
Біомаркери порушень скоротливої функції міокарда
Простеатозні біомаркери діабетичної кардіоміопатії
Маркери ремоделювання позаклітинного матриксу
Маркери фіброзу та запальних процесів
- Gilca G.E., Stefanescu G., Badulescu O., Tanase D.M., Bararu I., Ciocoiu M. Diabetic cardiomyopathy: current approach and potential diagnostic and therapeutic targets. J. Diabetes Res. 2017. 2017. 1310265. doi: 10.1155/2017/1310265.
- Ostanko V.L., Kalacheva T.P., Kalyuzhina E.V., Livshits I.K., Shalovay A.A., Chernogoryuk G.E. et al. Biological markers in risk stratification and progression of cardiovascular disease: present and future. Bull. Siberian Med. 2018. 17(4). 264-80. doi: 10.20538/1682-0363-2018-4-264-280. (in Russian)
- Jia G., Hill M.A., Sowers J.R. Diabetic cardiomyopathy: An update of mechanisms contributing to this clinical entity. Circ. Res. 2018. 122(4). 624-38. doi: 10.1161/CIRCRESAHA.117.311586.
- Lorenzo-Almorós A., Tuñón J., Orejas M., Cortes M., Egido J., Lorenzo O. Diagnostic approaches for diabetic cardiomyopathy. Cardiovasc. Diabetol. 2017. 16. 28. doi: 10.1186/s12933-017-0506-x.
- De Rosa S., Arcidiacono B., Chiefari E., Brunetti A., Indolfi C., Foti D.P. Type 2 Diabetes Mellitus and Cardiovascular Disease: Genetic and Epigenetic Links. Front Endocrinol (Lausanne). 2018. 9. 2. doi: 10.3389/fendo.2018.00002.
- Lee M.M.Y., McMurray J.J.V., Lorenzo-Almoros A., Kristensen S.L., Sattar N., Jhund P.S., Petrie M.C. Diabetic cardiomyopathy. Heart. 2019. 105(4). 337-45. doi: 10.1136/heartjnl-2016-310342.
- Chorna I., Motuziuk O. The characteristic of the main ischemic damaging biomarkers of muscle tissue. Lesia Ukrainka Eastern European National University Scientific Bulletin Series: Biol. Sciences. 2019. 387(3). 162-72. doi: 10.29038/2617-4723-2019-387-162-172. (in Ukrainian).
- Biological markers and their use in heart failure. Consensus of Ukrainian Association of Cardiology, Ukrainian Heart Failure Association and Ukrainian Association on Acute Cardiovascular Care. Ukrainian J. Cardiol. 2019. 26(2). 11-22. http://ucardioj.com.ua/index.php/UJC/article/view/168. (in Ukrainian)
- Ohkita M., Tawa M., Kitada K., Matsumura Y. Pathophysiological roles of endothelin receptors in cardiovascular diseases. J. Pharmacol. Sci. 2012. 119(4). 302-13. doi: 10.1254/jphs.12R01CR.
- Serhiyenko V.A., Serhiyenko A.A., Mankovsky B.N. Correlation between arterial wall stiffness, N-terminal prohormone of brain natriuretic peptide, functional and structural myocardial abnormalities in patients with type 2 diabetes mellitus and cardiac autonomic neuropathy. Diabetes mellitus. 2013. 16(4). 72-7. doi: 10.14341/DM2013472-77. (in Russian)
- D’Alessandro R., Masarone D., Buono A., Gravino R., Rea A., Salerno G. et al. Natriuretic peptides: molecular biology, pathophysiology and clinical implications for the cardiologist. Future Cardiol. 2013. 9(4). 519-34. doi: 10.2217/fca.13.32.
- Inoue Y., Kawai M., Minai K., Ogawa K., Nagoshi T., Ogawa T., Yoshimura M. The impact of an inverse correlation between plasma B-type natriuretic peptide levels and insulin resistance on the diabetic condition in patients with heart failure. Metab. Clin. Exp. 2016. 65(3). 38-47. doi: 10.1016/j.metabol.2015.09.019.
- Dencker M., Stagmo M., Dorkhan M. Relationship between natriuretic peptides and echocardiography parameters in patients with poorly regulated type 2 diabetes. Vasc. Health Risk Manag. 2010. 6. 373-82. doi: 10.2147/vhrm.s9332.
- Kiencke S., Handschin R., von Dahlen R., Muser J., Brunner-Larocca H.P., Schumann J. et al. Pre-clinical diabetic cardiomyopathy: prevalence, screening, and outcome. Eur. J. Heart Fail. 2010. 12(9). 951-7. doi: 10.1093/eurjhf/hfq110.
- Nunes S., Soares E., Fernandes J., Viana S., Carvalho E., Pereira F.C., Reis F. Early cardiac changes in a rat model of prediabetes: brain natriuretic peptide overexpression seems to be the best marker. Cardiovasc. Diabetol. 2013. 12. 44. doi: 10.1186/1475-2840-12-44.
- Korkmaz-Icoz S., Lehner A., Li S., Vater A., Radovits T., Brune M., Ruppert M. Left ventricular pressure-volume measurements and myocardial gene expression profile in type 2 diabetic Goto-Kakizaki rats. Am. J. Physiol. Heart Circ. Physiol. 2016. 311(4). 958-71. doi: 10.1152/ajpheart.00956.2015.
- Ruiz-Hurtado G., Gomez-Hurtado N., Fernandez-Velasco M., Calderon E., Smani T., Ordonez A. et al. Cardiotrophin-1 induces sarcoplasmic reticulum Ca(2+) leak and arrhythmogenesis in adult rat ventricular myocytes. Cardiovasc. Res. 2012. 96(1). 81-9. doi: 10.1093/cvr/cvs234.
- Gamela-Pozuelo L., Fuentes-Calvo I., Gomez-Marcos M.A., Recio-Rodriquez J.I., Agudo-Conde C., Fernandez-Martin J.L. et al. Plasma cardiotrophin-1 as a marker of hypertension and diabetes-induced target organ damage and cardiovascular risk. Medicine. 2015. 94(30). e1218. doi: 10.1097/md.0000000000001218.
- Garcia-Cenador M.B., Lopez-Novoa J.M., Diez J., Garcia-Criado F.J. Effects and mechanism of organ protection by cardiotrophin-1. Curr. Med. Chem. 2013. 20(2). 246-56. doi: 10.2174/0929867311320020005.
- Moreno-Aliaga M.J., Romero-Lozano M.A., Castano D., Prieto J., Bustos M. Role of cardiotrophin-1 in obesity and insulin resistance. Adipocyte. 2012. 1(2). 112-5. doi: 10.4161/adip.19696.
- Gamella-Pozuelo L., Fuentes-Calvo I., Gomez-Marcos M.A., Recio-Rodriguez J.I., Agudo-Conde C., Fernanez-Martin J.L. et al. Plasma cardiotrophin-1 as a marker of hypertension and diabetes-induced target organ damage and cardiovascular risk. Medicine. 2015. 94(30). e1218. doi: 10.1097/md.0000000000001218.
- Hung H.C., Lu F.H., Ou H.Y., Wu H.T., Wu J.S., Yang Y.C., Chang C.J. Increased cardiotrophin-1 in subjects with impaired glucose tolerance and newly diagnosed diabetes. Int. J. Cardiol. 2013. 169(3). e33-4. doi: 10.1016/j.ijcard.2013.08.112.
- Russell N.E., Higgins M.F., Amaruso M., Foley M., McAuliffe F.M. Troponin T and pro-B-type natriuretic peptide in fetuses of type 1 diabetic mothers. Diabetes Care. 2009. 32(11). 2050-5. doi: 10.2337/dc09-0552.
- Nyman K., Graner M., Pentikainen M.O., Lundbom J., Hakkarainen A., Siren R. et al. Cardiac steatosis and left ventricular function in men with metabolic syndrome. J. Cardiovasc. Magn. Reson. 2013. 15. 103. doi: 10.1186/1532-429X-15-103.
- Hoffmann U., Espeter F., Wei C., Ahmad-Nejad P., Lang S., Brueckmann M. et al. Ischemic biomarker heart-type fatty acid binding protein (hFABP) in acute heart failure-diagnostic and prognostic insights compared to NT-proBNP and troponin I. BMC Cardiovasc. Disord. 2015. 15. 50. doi: 10.1186/s12872-015-0026-0.
- Garcia-Rua V., Otero M.F., Lear P.V., Rodriguez-Penas D., Feijoo-Bandin S., Noguera-Moreno T. et al. Increased expression of fatty-acid and calcium metabolism genes in failing human heart. PLoS One. 2012. 7. e37505. doi: 10.1371/journal.pone.0037505.
- Fosshaug L.E., Dahl C.P., Risnes I., Bohov P., Berge R.K., Nymo S. et al. Altered levels of fatty acids and inflammatory and metabolic mediators in epicardial adipose tissue in patients with systolic heart failure. J. Card. Fail. 2015. 21(11). 916-23. doi: 10.1016/j.cardfail.2015.07.014.
- Ziyrek M., Kahraman S., Ozdemir E., Dogan A. Metformin monotherapy significantly decreases epicardial adipose tissue thickness in newly diagnosed type 2 diabetes patients. Rev. Port. Cardiol. 2019. 38(6). 419-23. doi: 10.1016/j.repc.2018.08.010.
- Wang T.D., Lee W.J., Shih F.Y., Huang C.H., Chen W.J., Lee Y.T. et al. Association of epicardial adipose tissue with coronary atherosclerosis is region-specific and independent of conventional risk factors and intra-abdominal adiposity. Atherosclerosis. 2010. 213(1). 279-87. doi: 10.1016/j.atherosclerosis.2010.07.055.
- Greulich S., de Wiza D.H., Preilowski S., Ding Z., Mueller H., Langin D. et al. Secretory products of guinea pig epicardial fat induce insulin resistance and impair primary adult rat cardiomyocyte function. J. Cell Mol. Med. 2011. 15(11). 2399-410. doi: 10.1111/j.1582-4934.2010.01232.x.
- Blumensatt M., Wronkowitz N., Wisa C., Cramer A., Mueller H., Rabelink M.J. et al. Adipocyte-derived factors impair insulin signaling in differentiated human vascular smooth muscle cells via the upregulation of miR-143. Biochim. Biophys. Acta Mol. Basis Dis. 2014. 1842(2). 275-83. doi: 10.1016/j.bbadis.2013.12.001.
- Chen W.J.Y., Greulich S., van der Meer R.W., Rijzewijk L.J., Lamb H.J., de Roos A. et al. Activin A is associated with impaired myocardial glucose metabolism and left ventricular remodeling in patients with uncomplicated type 2 diabetes. Cardiovasc. Diabetol. 2013. 12. 150(2013). doi: 10.1186/1475-2840-12-150.
- Mahabadi A.A., Berg M.H., Lehmann N., Kälsch H., Bauer M., Kara K. et al. Association of epicardial fat with cardiovascular risk factors and incident myocardial infarction in the general population: the Heinz Nixdorf Recall Study. J. Am. Coll. Cardiol. 2013. 61(13). 1388-95. doi: 10.1016/j.jacc.2012.11.062.
- Cavalcante J.L., Tamarappoo B.K., Hachamovitch R., Kwon D.H., Alraies M.C., Halliburton S. et al. Association of epicardial fat, hypertension, subclinical coronary artery disease, and metabolic syndrome with left ventricular diastolic dysfunction. Am. J. Cardiol. 2012. 110(12). 1793-8. doi: 10.1016/j.amjcard.2012.07.045.
- Abazid R.M., Smettei O.A., Kattea M.O., Sayed S., Saqqah H., Widyan A.M., Opolski M.P. Relation between epicardial fat and subclinical atherosclerosis in asymptomatic individuals. J. Thorac. Imaging. 2017. 32(6). 378-82. doi:10.1097/RTI.0000000000000296.
- Wu F.Z., Chou K.J., Huang Y.L., Wu M.T. The relation of locationspecific epicardial adipose tissue thickness and obstructive coronary artery disease: systemic review and meta-analysis of observational studies. BMC Cardiovasc. Disord. 2014. 14. 62. doi: 10.1186/1471-2261-14-62.
- Nerlekar N., Muthalaly R.G., Wong N., Thakur U., Wong D.T.L., Brown A.J., Marwick T.H. Association of volumetric epicardial adipose tissue quantification and cardiac structure and function. J. Am. Heart Assoc. 2018. 7(23). e009975. doi: 10.1161/JAHA.118.009975.
- Vrselja Z., Šram M., Andrijevic D., Takac B., Leksan I., Radic R., Curic G. Transcardial gradient of adiponectin, interleukin-6 and tumor necrosis factor-α in overweight coronary artery disease patients. Cytokine. 2015. 76(2). 321-7. doi: 10.1016/j.cyto.2015.09.009.
- Fitzgibbons T.P., Czech M.P. Epicardial and perivascular adipose tissues and their influence on cardiovascular disease: basic mechanisms and clinical associations. J. Am. Heart Assoc. 2014. 3(2). e000582. doi: 10.1161/JAHA.113.000582.
- Shimabukuro M., Hirata Y., Tabata M., Dagvasumberel M., Sato H., Kurobe H. et al. Epicardial adipose tissue volume and adipocytokine imbalance are strongly linked to human coronary atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2013. 33(5). 1077-84. doi: 10.1161/ATVBAHA.112.300829.
- Psaltis P.J., Talman A.H., Munnur K., Cameron J.D., Ko B.S.H., Meredith I.T. et al. Relationship between epicardial fat and quantitative coronary artery plaque progression: insights from computer tomography coronary angiography. Int. J. Cardiovasc. Imaging. 2016. 32(2). 317-28. doi: 10.1007/s10554-015-0762-3.
- Koval S.M., Yushko K.O., Snihurska I.O., Starchenko T.G., Pankiv V.I., Lytvynova O.M., Mysnychenko O.V. Relations of angiotensin-(1-7) with hemodynamic and cardiac structural and functional parameters in patients with hypertension and type 2 diabetes. Arterial Hypertension (Poland) 2019. 23(3). 183-189. doi: 10.5603/AH.a2019.0012.
- Sokolova L.K., Belchina Yu.V., Pushkarev V.V., Cherviakova S.A., Vatseba T.S., Kovzun O.I. et al. The blood level of endothelin-1 in diabetic patients depending on the characterisric of the disease. Mìžnarodnij endokrinologìčnij žurnal. 2020. 16(3). 35-45. doi: 10.22141/2224-0721.16.3.2020.205267.
- Ban C.R., Twigg S.M., Franjic B., Brooks B.A., Celermajer D., Yue D.K., McLennan S.V. Serum MMP-7 is increased in diabetic renal disease and diabetic diastolic dysfunction. Diabetes Res. Clin. Pract. 2010. 87(3). 335-41. doi: 10.1016/j.diabres.2010.01.004.
- Zaslavskaya E.L., Morozov A.N., Ionin V.A., Ma I., Nifontov S.Е., Baranova Е.I. et al. The role of transforming growth factor beta-1 and galectin-3 in formation of the left atrium fibrosis in patients with paroxysmal atrial fibrillation and metabolic syndrome. Russ. J. Cardiol. 2018. 154(2). 60-6. doi: 10.15829/1560-4071-2018-2-60-66. (in Russian)
- Tan S.M., Zhang Y., Wang B., Tan C.Y.R., Zammit S.C., Williams S.J. et al. FT23, an orally active antifibrotic compound, attenuates structural and functional abnormalities in an experimental model of diabetic cardiomyopathy. Clin. Exp. Pharmacol. Physiol. 2012. 39(8). 650-6. doi: 10.1111/j.1440-1681.2012.05726.x.
- Biernacka A., Cavalera M., Wang J., Russo I., Shinde A., Kong P. et al. Smad3 signaling promotes fibrosis while preserving cardiac and aortic geometry in obese diabetic mice. Circ. Heart Fail. 2015. 8(4). 788-98. doi: 10.1161/CIRCHEARTFAILURE.114.001963.
- Hutchinson K.R., Lord C.K., West T.A., Stewart J.A. Cardiac fibroblast-dependent extracellular matrix accumulation is associated with diastolic stiffness in type 2 diabetes. PLoS One. 2013. 8(8). e72080. doi: 10.1371/journal.pone.0072080.
- Shaver A., Nichols A., Thompson E., Mallick A., Payne K., Jones C. et al. Role of serum biomarkers in early detection of diabetic cardiomyopathy in the West Virginian Population. Int. J. Med. Sci. 2016. 13(3). 161-8. doi: 10.7150/ijms.14141.
- Russo I., Frangogiannis N.G. Diabetes-associated cardiac fibrosis: cellular effectors, molecular mechanisms and therapeutic opportunities. J. Mol. Cell Cardiol. 2016. 90. 84-93. doi: 10.1016/j.yjmcc.2015.12.011.
- Musimkhan M.K., Berkinbaev S.F., Shanazarov N.A., Karabaeva R.Zh., Kisikova S.D. Review of diagnostic methods for predicting the course of heart failure in patients with coronary artery disease. Modern problems of science and education. 2018. 2. 1-13. doi: 10.17513/spno.27450. (in Russian)
- Menini S., Iacobini C., Blasetti-Fantauzzi C., Pesce C.M., Pugliese G. Role of galectin-3 in obesity and impaired glucose homeostasis. Oxid. Med. Cell Longev. 2016. 2016. 9618092. doi: 10.1155/2016/9618092.
- Flores-Ramirez R., Azpiri-Lopez J.R., Gonzalez-Gonzalez J.G., Ordaz-Farias A., Gonzalez Carrillo L.E., Carrizales Sepulveda E.F., Vera-Pineda R. Global longitudinal strain as a biomarker in diabetic cardiomyopathy. A comparative study with gal-3 in patients with preserved ejection fraction. Arch. Cardiol. Mex. 2017. 87(4). 278-85. doi: 10.1016/j.acmx.2016.06.002.
- He J., Yu Z. The role of Galectin-3 in cardiac remodeling. Cardiol. Plus. 2016. 1(3). 28-36. doi: 10.4103/2470-7511.248355.
- 2017 ACC/AHA/HFSA Focused Update of the 2013 ACCF/AHA Guideline for the Management of Heart Failure. A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. J. Am. Coll. Cardiol. 2017. 70(6). 776-803. doi: 10.1016/j.jacc.2017.04.025.
- Pascual-Figal D.A., Lax A., Perez-Martinez M.T., del Carmen Asensio-Lopez M., Sanchez-Mas J. & GREAT Network. Clinical relevance of sST2 in cardiac diseases. Clin. Chem. Lab. Med. 2016. 54(1). 29-35. doi: 10.1515/cclm-2015-0074.
- Rodrigues P.G., Leite-Moreira A.F., Falcão-Pires I. Myocardial reverse remodeling how far can we rewind? Am. J. Physiol. Heart Circ. Physiol. 2016. 310(11). H1402-22. doi: 10.1152/ajpheart.00696.2015.
- Alonso N., Lupón J., Barallat J., de Antonio M., Domingo M., Zamora E. et al. Impact of diabetes on the predictive value of heart failure biomarkers. Cardiovasc. Diabetol. 2016. 15(1). 151. doi: 10.1186/s12933-016-0470-x.
- Huynh K., Bernardo B.C., McMullen J.R., Ritchie R.H. Diabetic cardiomyopathy: mechanisms and new treatment strategies targeting antioxidant signaling pathways. Pharmacol. Ther. 2014. 142(3). 375-415. doi: org/10.1016/j.pharmthera.2014.01.003.
- Guo R., Nair S. Role of microRNA in diabetic cardiomyopathy: from mechanism to intervention. Biochim. Biophys. Acta. 2017. 1863(8). 2070-7. doi: 10.1016/j.bbadis.2017.03.013.