
Журнал «Медицина неотложных состояний» Том 19, №8, 2023
Вернуться к номеру
Клініко-фармакологічні аспекти переходу між антиаритмічними препаратами
Авторы: Бездітко Н.В. (1), Романенко С.В. (2), Цубанова Н.А. (3)
(1) - Інститут підвищення кваліфікації спеціалістів фармації Національного фармацевтичного університету, м. Харків, Україна
(2) - м. Дніпро, Україна
(3) - Львівська медична академія ім. А. Крупинського, м. Львів, Україна
Рубрики: Эндокринология
Разделы: Справочник специалиста
Версия для печати
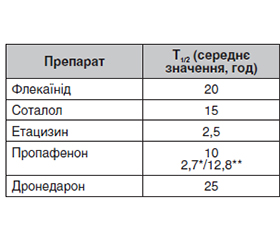
Серед когорти кардіологічних пацієнтів часті питання виникають при проведенні фармакотерапії аритмій. Після призначення антиаритмічного препарату (ААП) лікар може діагностувати недостатню ефективність терапії, розвиток побічних ефектів, що потребує негайної заміни препарату. Заміна одного антиаритміка на інший неможлива без урахування їх фармакокінетичних характеристик. У першу чергу слід мати на увазі такі показники, як період напіввиведення та особливості метаболізму системою цитохрому Р450. У статті актуалізовано інформацію щодо клініко-фармацевтичного профілю антиаритміків, що призначаються найбільш часто в Україні та світі. Саме фармакокінетичний профіль препарату дозволить зробити перехід на інший лікарський засіб максимально безпечним для пацієнта. Запропоновані практичні алгоритми переходу між ААП ґрунтуються на їхніх фармакокінетичних властивостях (розрахункові темпи елімінації) та притаманних тим чи іншим класам ААП електрокардіографічних критеріях потенційної проаритмогенної дії, оскільки при зміні антиаритмічних засобів важливо також моніторувати ЕКГ-критерії відсутності впливу залишкових концентрацій попереднього препарату. Подібний двофакторний підхід («відмивка» залежно від Т1/2 плюс ЕКГ-критерії) видається придатним для більшості антиаритмічних засобів.
Among the cohort of cardiac patients, the biggest problems arise during pharmacotherapy of arrhythmias. After prescribing an antiarrhythmic drug, the doctor may diagnose the insufficient effectiveness of the therapy, the development of side effects, which requires immediate replacement of the drug. It is impossible to replace one antiarrhythmic with another without considering their pharmacokinetic characteristics. First of all, such indicators as the half-life and peculiarities of metabolism by the cytochrome P450 system should be taken into account. The article updates information on the clinical and pharmaceutical profile of antiarrhythmics, which are prescribed most often in Ukraine and around the world. The pharmacokinetic characteristics of such antiarrhythmic drugs as flecainide, propafenone, etacizin, amiodarone, dronedarone, sotalol are given. It is the pharmacokinetic profile of the drug that will make switching to another drug safe for the patient. When replacing antiarrhythmic drugs, it is also advisable to monitor the ECG criteria to ensure the absence of the influence of residual concentrations of the previous drug. A similar two-factor approach (washout depending on Т1/2 plus ECG criteria) appears to be suitable for most antiarrhythmic drugs.
антиаритмічні препарати; період напіввиведення; ЕКГ-критерії; флекаїнід; цитохром Р450; метаболізм; безпека
antiarrhythmic drugs; half-life; ECG criteria; flecainide; cytochrome P450; metabolism; safety
/24.jpg)
Висновки
- Zhang J., Wang H., Fan Y., Yu Z., You G. Regulation of organic anion transporters: Role in physiology, pathophysiology, and drug elimination. Pharmacol. Ther. 2021 Jan. 217. 107647.
- Johnston A., Warrington S., Turner P. Flecainide pharmacokinetics in healthy volunteers: the influence of urinary pH. Br. J. Clin. Pharmacol. 1985 Oct. 20(4). 333-8.
- Conard G.J., Ober R.E. Metabolism of Flecainide. Am. J. Cardiol. 1984. 53. B41-B51.
- Holmes B., Heel R.C. Flecainide A Preliminary Review of Its Pharmacodynamic Properties and Therapeutic Efficacy. Drugs. 1985. 29. 1-33.
- Doki K. [Use of Pharmacogenetic Information for Therapeutic Drug Monitoring of an Antiarrhythmic Drug]. Yakugaku Zasshi. 2018. 138(9). 1145-1150. Japanese.
- Hanyok J.J. Clinical pharmacokinetics of sotalol. Am. J. Cardiol. 1993 Aug 12. 72(4). 19A-26A.
- Інструкція для застосування Соталол Сандоз; https:mozdocs.kiev.ua/likiview.php?id=28999
- Kimura M., Umemura K., Ikeda Y., Kosuge K., Mizuno A., Nakanomyo H., Ohashi K., Nakashima M. Pharmacokinetics and pharmacodynamics of (+/-)-sotalol in healthy male volunteers. Br. J. Clin. Pharmacol. 1996 Nov. 42(5). 583-8.
- da Cunha L.C., Gondim F.A., de Paola A.A., Barros I.C., Santos S.R. Kinetic disposition of (+)-S- and (-)-R-sotalol enantiomers in cardiac patients with tachyarrhythmias using an improved HPLC-fluorescence stereoselective method. Boll. Chim. Farm. 2002 Jan-Feb. 141(1). 45-51.
- Tjandramaga T.B., Verbeeck R., Thomas J., Verbesselt R., Verberckmoes R., Schepper P.J. The effect of end-stage renal failure and haemodialysis on the elimination kinetics of sotalol. Br. J. Clin. Pharmacol. 1976 Apr. 3(2). 259-65.
- Blair A.D., Burgess E.D., Maxwell B.M., Cutler R.E. Sotalol kinetics in renal insufficiency. Clin. Pharmacol. Ther. 1981 Apr. 29(4). 457-63.
- Hii J.T.Y., Duff H.J., Burgess E.D. Clinical Pharmacokinetics of Propafenone. Clin. Pharmacokinet. 1991. 21. 1-10.
- Інструкція для застосування Пропанорм® https:mozdocs.kiev.ua/likiview.php?id=5751.
- Tran Q.T., Baek I.H., Han N.Y., Yun H.Y., Chae J.W. The Effect of CYP2D6 Phenotypes on the Pharmacokinetics of Propafenone: A Systematic Review and Meta-Analysis. Pharmaceutics. 2022 Jul 11. 14(7). 1446.
- Lee J.T., Kroemer H.K., Silberstein D.J., Funck-Brentano C., Lineberry M.D., Wood A.J., Roden D.M., Woosley R.L. The role of genetically determined polymorphic drug metabolism in the beta-blockade produced by propafenone. N. Engl. J. Med. 1990 Jun 21. 322(25). 1764-8.
- Kroemer H.K., Fromm M.F., Bühl K., Terefe H., Blaschke G., Eichelbaum M. An enantiomer-enantiomer interaction of (S)- and (R)-propafenone modifies the effect of racemic drug therapy. Circulation. 1994 May. 89(5). 2396-400.
- Li G., Gong P.L., Qiu J., Zeng F.D., Klotz U. Stereoselective steady state disposition and action of propafenone in Chinese subjects. Br. J. Clin. Pharmacol. 1998 Nov. 46(5). 441-5.
- Zhou S.F. Polymorphism of human cytochrome P450 2D6 and its clinical significance: Part I. Clin. Pharmacokinet. 2009. 48. 689-723.
- Samer C.F., Lorenzini K.I., Rollason V., Daali Y., Desmeules J.A. Applications of CYP450 testing in the clinical setting. Mol. Diagn. Ther. 2013 Jun. 17(3). 165-84.
- Hindricks G., Potpara T., Dagres N., Arbelo E., Bax J.J., Blomström-Lundqvist C., et al.; ESC Scientific Document Group. 2020 ESC Guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European Association for Cardio-Thoracic Surgery (EACTS): The Task Force for the diagnosis and management of atrial fibrillation of the European Society of Cardiology (ESC) Developed with the special contribution of the European Heart Rhythm Association (EHRA) of the ESC. Eur Heart J. 2021 Feb 1. 42(5). 373-498. doi: 10.1093/eurheartj/ehaa612. Erratum in: Eur. Heart J. 2021 Feb 1. 42(5). 507. Erratum in: Eur. Heart J. 2021 Feb 1. 42(5). 546-547. Erratum in: Eur. Heart J. 2021 Oct 21. 42(40). 4194. PMID: 32860505.
- Белобородов В.Л., Бугрий Е.М., Залесская М.А., Тюкавкина Н.А., Каверина Н.В. Клиническая фармакокинетика этмозина и этацизина при их комбинированном применении. Хим.-фарм. журнал. 2004. Т. 38. № 2.
- Gneushev E.T., Ponomarenko E.L., Kurapov A.P., Kukes V.G. Farmakokinetika novogo otechestvennogo antiarutmicheskogo preparata étatsizina [Pharmacokinetics of the new Soviet anti-arrhythmia preparation etacizin]. Farmakol. Toksikol. 1984 Mar-Apr. 47(2). 52-4. Russian. PMID: 6714405.
- Kukes V.G., Shugushev Kh.Kh., Gneushev E.T., Ponomarenko E.L., Kurapov A.P. Farmakokinetika i farmakodinamika etatsizina [Pharmacokinetics and pharmacodynamics of ethacizin]. Sov. Med. 1986. (4). 81-2. Russian. PMID: 2424103.
- Reiffel J.A. Dronedarone: Where Does it Fit in the AF Therapeutic Armamentarium? J. Atr. Fibrillation. 2013 Apr 6. 5(6). 752.
- Rosa G.M., Bianco D., Parodi A., Valbusa A., Zawaideh C., Bizzarri N., et al. Pharmacokinetic and pharmacodynamic profile of dronedarone, a new antiarrhythmic agent for the treatment of atrial fibrillation. Expert Opin. Drug Metab. Toxicol. 2014 Dec. 10(12). 1751-64.
- Naccarelli G.V., Bhatt D.L., Camm A.J., Le Heuzey J.Y., Lombardi F., Tamargo J., et al.; ARTEMIS AF Investigators. Evaluation of the Switch from Amiodarone to Dronedarone in Patients with Atrial Fibrillation: Results of the ARTEMIS AF Studies. J. Cardiovasc. Pharmacol. Ther. 2020 Sep. 25(5). 425-437.
- Tamargo J., López-Farré A., Caballero R., Delpón E. Dronedarone. Drugs Today (Barc). 2011 Feb. 47(2). 109-33.
- Brenner R., Delacrétaz E. Dronedarone for the management of atrial fibrillation. Swiss Med. Wkly. 2011 Mar 2. 141. w13158.
- Safety of dronedarone (Multaq). Med. Lett. Drugs Ther. 2011 Dec 12. 53(1379-1380). 103-4. PMID: 22173456.
- Hassib M., Elkhouly A., Ansari S., Hamilton S., Kapaganti S. A Case of Dronedarone-Induced Hypotensive Shock. Cureus. 2020 Jun 6. 12(6). e8478.
- Epstein A.E., Olshansky B., Naccarelli G.V., Kennedy J.I. Jr, Murphy E.J., Goldschlager N. Practical Management Guide for Clinicians Who Treat Patients with Amiodarone. Am. J. Med. 2016. 129. 468-75.
- Immordino L., Connolly S., Crijns H., Roy D., Capucci A., Radzik D., et al. Effects of dronedarone started rapidly after amiodarone discontinuation. Clin. Cardiol. 2013 Feb. 36(2). 88-95.