
Журнал «Здоровье ребенка» 4 (25) 2010
Вернуться к номеру
Молекулярно-генетические механизмы развития и современные методы лечения легочной артериальной гипертензии у детей
Авторы: Волосовец А.П.1, Абатуров А.Е.2, Агафонова Е.А.2, 1 Национальный медицинский университет им. А.А. Богомольца, г. Киев, 2 Днепропетровская государственная медицинская академия
Рубрики: Педиатрия/Неонатология
Версия для печати
В обзоре представлены механизмы реализации влияния хронической гипоксии, воспаления, нарушений гемодинамики, ВИЧ и HHV-8 на развитие легочной артериальной гипертензии.
Легочная артериальная гипертензия, молекулярно-генетические механизмы, дети.
Сокращения: АКМ (активные кислородсодержащие метаболиты), ВИЧ (вирус иммунодефицита человека), ЛАГ (легочная артериальная гипертензия), ALK (активинподобная киназа), Angp-1 (ангиопоэтин-1), Angt (ангиотензин), AP-1 (активирующий протеин 1), BMP (костные морфогенетические белки), BMPR2 (рецептор II типа костного морфогенетического белка), CD36 (рецептор тромбоспондина), COX (циклооксигеназа), ELAM (эндотелиновая молекула адгезии лейкоцитов), ET (эндотелин), FOXO1 (forkhead transcription factor FKHR), HHV-8 (герпесвирус 8-го типа), HIF (индуцибельный гипоксией фактор), HSP (белок теплового шока, шаперон), ICAM-1/CD54 (межклеточная адгезивная молекула-1), ICAM-2/CD102 (межклеточная адгезивная молекула-2), IFN (интерферон), IP-10/CXCL10 (интерферон- g -индуцируемый протеин 10 kDa), LANA-1 (latency-associated nuclear antigen 1), LKLF/KLF2 (lung Kr ь ppel-like factor), MCP1/CCL2 (макрофагальный хемоаттрактантный протеин), MIP-1 a /CCL3 (макрофагальный воспалительный протеин 1 a ), MIP-1 b /CCL4 (макрофагальный воспалительный протеин-1 a ), MLCK (киназа легких цепей миозина), MMP (матриксная металлопротеиназа), NF-IL6 (нуклеарный фактор IL6), NF- k В (нуклеарный фактор k В), NO (монооксид азота), NOS (нитрооксидсинтаза), PDGF (тромбоцитарный фактор роста), PGI 2 (простациклин), RANTES/CCL5 (регулятор активации нормальной Т-клеточной экспрессии и секреции); ROC (receptor-operated cation channels/рецептор-управляемые каналы), SERT (серотониновый транспортер), SOC (store-operated channels/резерв-управляемые каналы), TGF- b (трансформирующий фактор роста b ), TIMP-1 (тканевой ингибитор металлопротеиназ), TRPC (transient receptor potential channels), TX A2 (тромбоксан A2), TXAS (тромбоксансинтаза), VCAM-1 (адгезивная молекула I сосудистого эндотелия), VEGF (сосудистый эндотелиальный фактор роста), VIP (вазоактивный интестинальный пептид), EDNR (эндотелиновый рецептор), MLCK (киназа легких цепей миозина), ИЛАГ (идиопатическая ЛАГ), FGF (фактор роста фибробластов), VGCC (вольтажзависимые Са 2+ -каналы), IQGAP1 (ключевой регулятор клеточно-клеточной адгезии), NOX4 (субъединица NADPH-оксидазы).
Введение
Инициирующими факторами развития легочной артериальной гипертензии у детей с генетической предрасположенностью могут выступать: воспаление; хроническая гипоксия; диффузные заболевания соединительной ткани; врожденные пороки сердца (системно-легочные шунты); портальная гипертензия; другие заболевания, сопровождающиеся постоянным изменением легочного кровотока; заболевания, протекающие с тромбоэмболией; ВИЧ-инфекция; герпесвирусная инфекция 8-го типа; влияние токсических веществ [1, 140].
Роль хронической гипоксии в развитии ЛАГ
Гипоксия регулирует множество генов, участвующих в регуляции гликолиза [103], липидного, белкового обменов [91, 93, 114]. Под влиянием гипоксии происходит выраженная вазоконстрикция, ремоделирование стенки сосудов легких, увеличивается коагуаляционный потенциал крови, индуцируется процесс воспаления.
Гипоксия в результате прямого и опосредованного действия кислородзависимых механизмов вызывает выраженную констрикцию сосудов, преимущественно прекапиллярного ложа легких (рис. 1) [35, 129, 226]. Ведущим механизмом вазоконстриктивного действия гипоксии является ингибиция экспрессии O 2 -чувствительных вольтажзависимых калиевых каналов Kv 1.1, Kv 1.2, Kv 1.5, Kv 2.1 и Kv 9.3 гладкомышечных клеток сосудов [109, 112, 134, 207], что приводит к деполяризации мембраны клетки и активации L -типа VDCC , которые обусловливают прохождение ионов Ca 2+ в клетку, вызывая вазоконстрикцию [25, 129, 167].
Гипоксия сопровождается образованием избытка АКМ, которые могут ингибировать основные вазодилатационные молекулярные механизмы, связанные с действием NO , PGI 2 , EDHF [67].
Хроническая гипоксия сопровождается изменением экспрессии множества генов эндотелиоцитов и гладкомышечных клеток, участвующих в регуляции тонуса сосудов [4, 166]. В частности, происходит снижение продукции вазодилатирующих факторов NO , PGI 2 , адреномедуллина, гемоксигеназы-1 и увеличение продукции ET, серотонина, T X A2 , сфингозина 1, экспрессии EDNR-1 В рецепторов, SERT, обусловливающих вазоконстрикцию [198]. Вазоконстрикторы, продукция которых вызвана гипоксией, индуцируют активность малых ГТФ-связанных протеинов ( Rho , Ras , Rab , Sarl / Arf , и Ran семейств), которые ингибируют миозиновую фосфатазу ( MLCPh ), что приводит к Ca 2+ /калмодулин-зависимой активации MLCK и сокращению гладкомышечных клеток [133, 203]. Гипоксия индуцирует активность PL А и PL С, диацилглицероллипазы, что сопровождается изменением липидного бислоя мембраны эндотелиоцитов и усилением продукции дериватов арахидоновой кислоты, которые непосредственно влияют на тонус сосудов [198].
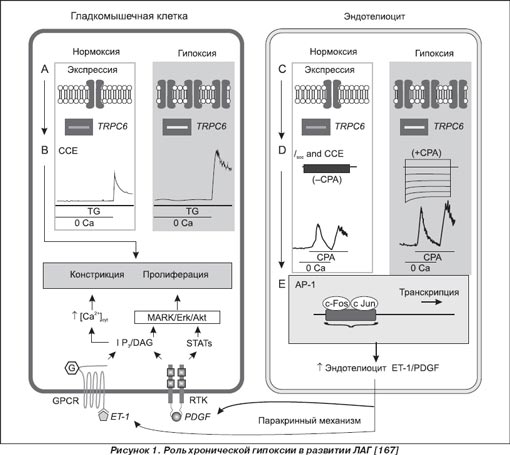
При дефиците кислорода снижается активность кислородзависимых реакций, в частности понижается уровень гидроксилирования пролиновых остатков (P 402 и P 564 ) в a -субъединице HIF-1. Гидроксилирование a -субъединицы HIF-1, которое является кислородзависимой реакцией, приводит к ее полиубиквитинированию и протеасомной деградации. Дополнительная активация HIF-1 происходит за счет еще одной системы сенсинга концентрации кислорода: малых ГТФаз семейства Rho [27]. Негидроксилированная a -субъединица HIF-1 формирует с конститутивно экспрессируюшейся b -субъединицей HIF-1 супрамолекулярный комплекс, который, присоединяясь к соответственным cis -элементам ДНК, регулирует экспрессию более 100 генов, необходимых для адаптации клеток к гипоксии [27, 106, 117]. В частности, генов, участвующих в эритропоэзе и обмене железа — эритропоэтина, трансферина, трансферинового рецептора, церулоплазмина; в ангиогенезе – TGF b 3 , VEGF , EG - VEGF , MMP2, катепсина D ; в регуляции сосудистого тонуса — iNOS , ET -1, адреномедуллина, a 1в -адренорецептора; в регуляции метаболизма глюкозы — аденилаткиназы-3, альдолазы-А, -С, карбоангидразы-9, энолазы-1, транспортеров глюкозы 1 и 3, гексокиназы 1 и 2, лактатдегидрогеназы-А, пируваткиназы-М, фосфофруктокиназы L , активирующих анаэробное дыхание; в регуляции клеточной пролиферации и выживания — TGF , адреномедуллина; в регуляции апоптоза — протеина BNip 3, Nix . Однако многие из данных молекулярных факторов участвуют в процессах вазоконстрикции и пролиферации гладкомышечных клеток сосудов легких [42, 182, 183, 185, 222, 233]. HIF-2 a в отличие от полиубиквитарного HIF-1 a экспрессирован только в гладкомышечных клетках сосудов легких и клетках, продуцирующих катехоламины, и индуцирует преимущественно продукцию ET -1 [148, 178, 183, 184].
Образующиеся в результате гипоксии АКМ принимают участие в индуцированной гипоксией вазоконстрикции и пролиферации гладкомышечных клеток. Под влиянием гипоксии происходит увеличение продукции TGF- b 1 , который индуцирует экспрессию NOX4 — субъединицы NADPH-оксидазы. АКМ, обусловленные активностью NOX4, индуцируют синтез индуцибельных гипоксией факторов — HIF-1 a , HIF-2 a , HIF-3 a [118, 178, 183, 227].
Гипоксия подавляет продукцию эндотелиоцитами тромбомодулина и увеличивает экспрессию тканевого фактора, повышая прокоагуляционную активность [200].
Под влиянием гипоксии происходит активация факторов транскрипции NF - k В, NF - IL 6, что приводит к усилению продукции: 1) провоспалительных цитокинов IL -1 b , IL -2, IL -6, IL -12, IL -18, TNF - a , TNF - b , гранулоцитарного колониестимулирующего фактора, гранулоцитарно-макрофагального колониестимулирующего фактора, IFN - b , IFN - g ; 2) хемокинов — GRO a , GRO b , GRO g , IL -8, IP -10/ CXCL 10, MIP -1 a / CCL 3, MIP -1 b , RANTES / CCL 5; 3) адгезинов — Е-селектина, ICAM -1/ CD 54, ICAM -2, CD 102, VCAM -1, ELAM , MACAM -1; 4) острофазовых белков — С-реактивного протеина, сывороточного амилоид-А-протеина; В, С3, С4 факторов комплемента, липополисахаридсвязывающего белка, кислого a 1-гликопротеина; 5) иммунорегуляторных молекул — k легких цепей иммуноглобулинов, инвариантных цепей; 6) костимулирующих молекул CD 80, CD 86 на антигенпрезентирующих клетках; 7) антигенов I и II класса HLA системы; 8) b 2-микроглобулина 9) a - и b -рецепторов T -клеток; 10) транспортера, ассоциированного с процессингом антигена; 11) индуцибельных ферментов нитрооксидсинтазы, COX-2, PLА 2 ; 12) ММР и др. [116, 139, 198].
Хроническая гипоксия приводит к ремоделированию сосудов [164]. В условиях гипоксии увеличивается продукция эндотелиальными клетками ламинина, фибронектина, эластина, значительно снижается синтез протеингликанов и молекул, которые ингибируют рост гладкомышечных клеток. Данные изменения в спектре эндотелиальной продукции обусловливают усиленную пролиферацию гладкомышечных клеток. Гипоксия активирует резидентные адвентициальные фибробласты, вызывая продукцию провоспалительных цитокинов и хемокинов, которые привлекают моноциты/фиброциты к периваскулярному пространству, оказывая прямое действие на процессы ремоделирования, ангиогенеза, синтез коллагена и др. [17, 146, 198]. Гладкомышечные клетки сосудов являются высокопластичными клетками, которые, изменяя под влиянием экзогенных факторов экспрессию генов, достаточно быстро меняют свой статус — от контрактильного к мигрирующему или пролиферирующему. Известно, что контрактильность и фенотипические метаморфозы гладкомышечных клеток зависят от изменения внутриклеточной концентрации ионов Са 2+ [224]. Основными системами, регулирующими уровень цитозольного Са 2+ , являются вольтажзависимые Са 2+ -каналы (VGCC); вольтажнезависимые Са 2+ -каналы — TRP C и ROC, SOC, которые также состоят из субъединиц TRP C ; механизмы высвобождения и секвестрации Са 2+ саркоплазматическим и эндоплазматическим ретикулумом; высвобождающая из клетки во внеклеточное пространство Ca 2+ -Mg 2+ АТФазная помпа; Na + -Ca 2+ обменник, митохондриальные и лизосомальные механизмы высвобождения и секвестрации Са 2+ [5, 72, 210, 211, 225]. Показано, что гиперэкспрессия TRPC6 приводит к пролиферации гладкомышечных клеток [61], а в развитии неоинтимы принимают участие TRPC1 [155].
Под влиянием гипоксии увеличивается продукция компонентов экстрацеллюлярного матрикса — коллагенов 1, 3, 4 и 18, MMP-2, виментина и тенасцина C, на фоне снижения синтеза протеингликана декорина, который является ингибитором TGF- b и PDGF, a -катенина и IQGAP1 — ключевого регулятора клеточно-клеточной адгезии [41, 84, 213]. Тенасцины — высококонсервативное семейство больших олигомерических гликопротеинов экстрацеллюлярного матрикса, молекулы которых имеют повторяемые последовательности, подобные повторам молекул эпителиального фактора роста, фибронектина, фибриногена [95]. Показано, что тенасцин C, как мощный митоген, индуцирует пролиферацию гладкомышечных клеток и может вызвать трансформацию мезенхимальных клеток в эндотелиоциты, тем самым привести к организации неоинтимы [82, 137, 163].
Также под действием гипоксии в гладкомышечных клетках и фибробластах легочной ткани усиливается экспрессия FK 506-связывающего протеина 1 a (12 kDa ), просаросина, FISP , альдолазы 3 C , S 100 A 4, CD 36 FKBP 1 a и подавляется экспрессия остеоглицина, гомолога 10-го клеточного цикла, HSP 60, протеина, связывающего клеточную нуклеиновую кислоту [66].
Роль воспаления в развитии ЛАГ
Показано, что даже «чистые» формы ИЛАГ сопровождаются повышенным уровнем в сыворотке крови IL-1 и IL-6, которые являются мощными митогенами и способствуют развитию тромбоза. Повышение давления в легочных артериях и хроническая гипоксия сопровождаются увеличением концентрации солютабного CD40, MCP1/CCL2, хемоаттрактанта для моноцитов и T-клеток RANTES/CCL5, фракталкина/CX3CL1, обусловливая возникновение воспалительного процесса в стенках мелких легочных артерий [196, 212]. Мембранный фракталкин, продуцируемый эндотелиальными клетками, играет важную роль в адгезии клеток крови, несущих фракталкиновый рецептор CX3CR1 (моноцитов, натуральных киллеров, цитотоксических Т-лимфоцитов) [76, 105]. CCL 2 и фракталкин способствуют пролиферации гладкомышечных клеток в мелких сосудах легких [99].
Но и сам процесс хронического воспаления может быть решающим фактором развития легочной гипертензии [45].
Неаддитивная роль процессов хронической гипоксии и воспаления в развитии ЛАГ
Взаимодействие двух ведущих факторов развития ЛАГ — хронической гипоксии и воспаления — особенно показательно на примере системы Angp -1. Ангиопоэтин-1 (70- kD ) является ангиогенным фактором роста и играет центральную роль в ангиогенезе [122]. Баланс и последовательная экспрессия Ang p -1 и VEGF необходимы для формирования новых сосудов. Ang p -1 является агонистом рецептора тирозиновой киназы Tie2. Продукция Ang p -1 и экспрессия рецептора Tie2 индуцируются гипоксией и регулируются IL-1 b , VEGF, TGF- b , TNF- a (табл. 1) [122, 193].
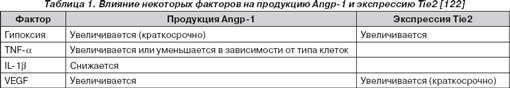
Ang p -1 и рецептор Tie2 играют важнейшую роль в развитии ЛАГ. Отмечено, что при ЛАГ значительно увеличивается продукция Ang p -1. Гиперпродукция Ang p -1 является причинным фактором ЛАГ, а не следствием ее развития [140]. Хроническое воспаление бронхолегочных структур, которое сопровождается усилением продукции TNF- a , обусловливает индукцию синтеза Ang p -1, а гипоксия — как индукцию синтеза Ang p -1, так и экспрессию рецепторов Tie2. Рецепторы Tie2 преимущественно экспрессируются на клеточных мембранах эндотелиоцитов. Но их экспрессия характерна и для эпителиоцитов, гладкомышечных клеток, фибробластов, моноцитов, нейтрофилов, эозинофилов. Возбуждение рецептора Tie2 вызывает хемотаксис гладкомышечных клеток, нейтрофилов, эозинофилов, индуцирует дифференцировку мезенхимальных клеток в гладкомышечные клетки. Таким образом, усиленная продукция Ang p -1, возбуждение рецепторов Tie2 обусловливают гиперпролиферацию гладкомышечных клеток. Индукция рецепторов Tie2 также активирует продукцию серотонина, MMP, плазмина и ингибирует продукцию TIMP [108, 122, 193, 206]. У пациентов с ИЛАГ Angp -1 увеличивает экспрессию ET-1 и серотонина [12, 168]. Протеин Angp -1 обладает антиапоптотическим действием на эндотелиальные клетки, ингибируя фактор транскрипции FOXO 1 через активацию Akt [13]. Angp -1 нарушает эффективность передачи сигналов от BMPR - II и резко снижает внутриклеточный уровень концентрации мРНК BMPR - IA в эндотелиоцитах [140]. По всей вероятности, ингибирующее влияние Angp -1 на активность BMPR - II , BMPR - IA является одним из связующих звеньев патогенезов спорадической и семейной легочной гипертензии [194].
Роль нарушений гемодинамики в развитии ЛАГ
Изменения скорости движения и давления крови в легочных сосудах приводит к изменению положения прореснички эндотелиоцита [24]. Первичный изгиб прореснички приводит опосредовано через полицистин-2 к повышению уровня внутриклеточной концентрации ионов Са 2+ [158]. Изменения положения прореснички изменяет состояние цитоскелета клетки, [37, 73, 125, 195], активирует механорецепторы (интегриновые рецепторы, G-протеиновые рецепторы, ионные каналы), что обусловливает изменение экспрессии множества генов, в том числе вазоактивных медиаторов (ET-1, Ang t II, тромбоксанов, eNOS , LKLF / KLF 2, PGI 2 ), факторов роста ( PDGF , FGF , VEGF , TGF - b ) [2, 50, 74, 113, 157]. Усиление продукции KLF2 (члена SP/XKLF-семейства факторов транскрипции цинкового пальца Cys2/His2) сопровождается индукцией экспрессии eNOS [111, 153]. Под влиянием механосенситивных рецепторов увеличивается экспрессия TRPM 7 на мембране гладкомышечных клеток, способствуя их пролиферации [143].
Роль инфекционных агентов в развитии ЛАГ
Экзогенными факторами, увеличивающими риск развития ЛАГ, являются определенные вирусные инфекции (ВИЧ, HHV-8) [177].
Нарушение функционирования BMPR2 может быть вызвано не только мутациями. Так, показано, что белок tat ВИЧ-1 ингибирует экспрессию BMPR 2 [26] . В последние годы появились доказательства участия HHV-8, обладающего эндотелиотропностью, в развитии апоптозрезистентных клонов эндотелиоцитов и плексиформных поражений. Инфекционный процесс, вызванный HHV-8, сопровождается подавлением продукции BMP4, высокой экспрессией вирусных циклина и LANA-1. Структура вирусного циклина гомологична структуре циклина D, который направляет циклинзависимые киназы 4 и 6 к белку ретинобластомы. Фосфорилированный белок ретинобластомы участвует в регуляции гена p14ARF и генов, определяющих клеточный цикл. Вирусный циклин, конкурируя с циклином D , ингибирует активность белка ретинобластомы, что ведет к ускорению клеточного цикла и ингибиции экспрессии p14ARF. Дефицит p14ARF приводит к снижению экспрессии р53, тем самым ингибируя р53-зависимый апоптоз. Протеин LANA-1 также подавляет активность р53. Таким образом, латентная HHV-8 инфекция может привести к пролиферации апоптозрезистентных клонов эндотелиоцитов, уменьшению просвета мелких сосудов и, как следствие, легочной гипертензии [34, 65, 96].
Список литературы находится в редакции