
Международный эндокринологический журнал 2 (66) 2015
Вернуться к номеру
Спектр клініко-патогенетичних варіантів первинного гіперальдостеронізму
Авторы: Щекатурова Л.В. — Український науково-практичний центр ендокринної хірургії, трансплантації ендокринних органів і тканин МОЗ України, м. Київ
Рубрики: Эндокринология
Разделы: Справочник специалиста
Версия для печати
У роботі проведено дослідження структури окремих клініко-патогенетичних варіантів первинного гіперальдостеронізму на матеріалі клініки Українського науково-практичного центру ендокринної хірургії, трансплантації ендокринних органів і тканин. Було ретроспективно досліджено дані 474 хворих, які підлягали хірургічному та консервативному лікуванню протягом 1996–2014 рр. Усім хворим проводили скринінгове дослідження калію крові, альдостерон-ренінового співвідношення, мультиспіральну комп’ютерну томографію, підтверджувальні тести; 63 хворим було проведено роздільний забір крові з надниркових вен і нижньої порожнистої вени. Аналіз структури окремих клініко-патогенетичних форм дозволив виділити такі основні групи: двобічну ідіопатичну гіперплазію надниркових залоз — 235 (49,6 %), альдостеронпродукуючу аденому — 195 (41,1 %), первинну (однобічну) гіперплазію надниркових залоз — 36 (7,6 %), сімейні форми — 6 (1,3 %) (у т.ч. глюкокортикоїдчутливий альдостеронізм — 4, FH II — 2), адренокортикальний рак — 2 (0,4 %). Прооперовано 255 хворих з первинним гіперальдостеронізмом шляхом однобічної лапароскопічної адреналектомії. Із 219 хворих групи консервативного лікування в 22 пацієнтів було запідозрено глюкокортикоїдчутливу форму первинного гіперальдостеронізму, проведено пробне лікування дексаметазоном, що підтвердило діагноз у чотирьох пацієнтів, які з успіхом продовжили лікування цим препаратом у дозі 0,25–0,5 мг на добу. Решта пацієнтів отримували консервативне лікування із застосуванням блокаторів мінералокортикоїдних рецепторів. Показано, що своєчасне визначення форми захворювання дозволяє вибрати адекватний спосіб лікування.
В работе исследована структура отдельных клинико-патогенетических вариантов первичного гиперальдостеронизма по материалам клиники Украинского научно-практического центра эндокринной хирургии, трансплантации эндокринных органов и тканей. Ретроспективно были исследованы данные 474 больных, которые подвергались хирургическому и консервативному лечению на протяжении 1996–2014 гг. Всем пациентам проводили скрининговое исследование калия крови, альдостерон-ренинового соотношения, мультиспиральную компьютерную томографию, подтверждающие тесты; 63 больным был проведен раздельный забор крови из надпочечниковых вен и нижней полой вены. Анализ структуры отдельных клинико-патогенетических форм дал возможность выделить следующие основные группы: двухстороннюю идиопатическую гиперплазию надпочечниковых желез — 235 (49,6 %), альдостеронпродуцирующую аденому — 195 (41,1%), первичную (одностороннюю) гиперплазию надпочечниковых желез — 36 (7,6 %), семейные формы — 6 (1,3 %) (в т.ч. глюкокортикоидчувствительній альдостеронизм — 4, FH II — 2), адренокортикальный рак — 2 (0,4 %). Прооперировано 255 больных с первичным гиперальдостеронизмом путем односторонней лапароскопической адреналэктомии. Из 219 пациентов группы консервативного лечения у 22 пациентов было подозрение на глюкокортикоидчувствительный вариант первичного гиперальдостеронизма. Было назначено пробное лечение дексаметазоном, которое подтвердило диагноз у 4 пациентов, которые успешно продолжили лечение данным препаратом в дозе 0,25–0,5 мг в сутки. Остальные пациенты получали консервативное лечение блокаторами минералокортикоидных рецепторов. Показано, что своевременное определение формы заболевания позволяет выбрать адекватный способ лечения.
The aim of the study was to investigate the spectrum of different pathogenetic forms of primary hyperaldosteronism on the clinical data of Ukrainian Scientific and Practical Center of Endocrine Surgery, Endocrine Organs and Tissues Transplantation within the period of 1996–2014 yrs. Results of diagnosis and treatment of 474 surgical and therapeutical patients were retrospectively studied. All patients were undergone screening of serum potassium, aldosteron-renin ratio, confirmation tests, multi-spiral computed tomography. Selective adrenal vein sampling was performed in 63 cases. Analysis of separate clinical and pathogenetic forms demonstrated the following sharing: idiopathic adrenal hyperplasia — 235 (49.6 %), aldosteron-producing adenoma — 195 (41.1 %), primary (unilateral) adrenal hyperplasia — 36 (7.6 %), familial forms — 6 (1.3 %) (including glucocorticoid-remediable aldosteronism — 4, FH II — 2), adrenocortical carcinoma — 2 (0.4 %). 255 patients with primary hyperaldosteronism were operated by unilateral laparoscopic adrenalectomy. Among 219 patients from the group of conservative treatment primary hyperaldosteronism glucocorticoid-remediable aldosteronism was suspected in 22 patients and was confirmed by presumptive treatment with dexamethasone in 4 patients who continued the successful treatment with this medication dosed 0.25–0.5 mg daily. The rest of patients received treatment with antagonists of mineralocorticoids receptors. It was clearly shown that in-time diagnosis of distinct form of primary hyperaldosteronism allows chose adequate mode of therapy.
первинний гіперальдостеронізм, патогенез, варіанти захворювання.
первичный гиперальдостеронизм, патогенез, варианты заболевания.
primary hyperaldosteronism, pathogenesis, variety of disease.
Статья опубликована на с. 132-137
Вступ
Первинний гіперальдостеронізм (ПГА) — ендокринне захворювання, яке характеризується автономною гіперпродукцією мінералокортикоїдного гормона альдостерону клітинами кіркової речовини надниркових залоз (НЗ). ПГА — найбільш поширене захворювання НЗ та найчастіша (90 %) з ендокринних причин артеріальної гіпертензії (АГ) [1–3, 5–9]. Водночас точкою впливу альдостерону є не тільки мінералокортикоїдні рецептори судин, але й серце, нирки, головний мозок, легені, кишечник, ушкодження яких може проявитися в інших, крім АГ, симптомах [3]. Специфічний руйнівний вплив спричиняється альдостероном через стимулювання фібробластичної активності в судинах та міокарді, пригнічення фібринолізу, накопичення колагену та гіпертрофію міокарда (особливо лівого шлуночка), а також ремоделювання судин з їх стенозом і склерозом. Причому наразі доведено (як в експерименті, так і в клініці) незалежний від артеріального тиску (АТ) вплив ПГА на лівошлуночкову гіпертрофію, а також можливість зворотного розвитку її під впливом дії блокаторів рецепторів альдостерону або після адреналектомії з приводу альдостеронпродукуючої аденоми НЗ [5, 8].
Серед усіх випадків артеріальних гіпертензій ПГА становить 6–15 %, у тому числі 3–5 % пацієнтів з АГ без гіпокаліємії [7–9]. З огляду на 30% поширеність АГ в популяції можна припустити не менше 1 мільйона потенційних пацієнтів в Україні.
Усе це свідчить про надзвичайно велику соціальну значущість проблеми виявлення та лікування ПГА, адже етіопатогенетично спрямоване лікування є набагато ефективнішим та дешевшим за емпіричні схеми комбінацій багатьох антигіпертензивних препаратів, які до того ж не здатні запобігти специфічним ускладненням тривалої дії надлишку альдостерону (гіпокаліємічні аритмії та нефропатії, лівошлуночкова гіпертрофія міокарда, ішемічні ушкодження мозку).
Водночас сам по собі ПГА є ендокринним захворюванням, що складається з декількох різних за етіологією та патологічною природою спорадичних і генетичних, пухлинних, гіперпластичних та ферментативних порушень НЗ. Причому розуміння природи ПГА еволюціонує від уявлення причиною захворювання спочатку виключно альдостеронпродукуючої аденоми НЗ, потім — різних типів аденом і гіперплазії кори НЗ, а останніми роками поступово покращилось розуміння генетичних передумов розвитку морфологічних та ферментативних порушень, що призводять до гіперальдостеронізму.
Окремі клініко-патогенетичні форми ПГА характеризуються особливостями пріоритетного застосування різних методів лікування. Саме тому не тільки донесення до відома про високу поширеність ПГА та методів діагностики цього захворювання, але й набуття досвіду у визначенні окремих патогенетичних форм цієї патології та їх диференційованого лікування залишається вкрай актуальним питанням.
Форми ПГА. ПГА може мати спорадичний чи сімейний характер. Основними причинами спорадичного варіанта є двобічна ідіопатична гіперплазія кори НЗ (ІГК) та альдостеронпродукуюча аденома (АПА) — переважно невеличка пухлина діаметром 0,5–3 см, часто множинна в межах однієї залози. Цікаво, що цим двом основним варіантам ПГА притаманне походження з епітеліальних клітин сітчастої зони кори НЗ. Окремо виділяють доволі рідкісну форму альдостеронпродукуючої аденоми, що чутлива до реніну (РЧАПА).
Менш поширеним варіантом є однобічна гіперплазія клубочкової зони кори НЗ (так звана первинна гіперплазія НЗ, ПГНЗ). Характерною морфологічною рисою РЧАПА та ПГНЗ є походження з клітин гломерулярної зони кори НЗ, що може бути основою гістологічного підтвердження цих різновидів ПГА. Рідкісною причиною ПГА може бути карцинома кори НЗ; її слід підозрювати при розмірі, більшому за 3 см. Казуїстичним джерелом надлишкового синтезу альдостерону може бути ектопічна його секреція пухлинами яєчників та нирок.
Сімейна форма ПГА охоплює лише 2–5 % випадків хвороби, є генетично детермінованою автосомно-домінантною патологією та має три типи (FH I, FH IІ та FH IIІ), перший з яких має назву глюкокортикоїдчутливого альдостеронізму (ГКЧА). Вона є наслідком утворення гена CYP11B1/CYP11B2, який кодує синтез 11-гідроксилази та альдостеронсинтетази, що стимулюються адренокортикотропним гормоном (АКТГ). Ця форма захворювання, відома з 1966 року, може успішно лікуватися глюкокортикоїдами. Інша сімейна форма (FH II), вперше описана в 1991 р., не реагує на лікування глюкокортикоїдами. Вона може демонструвати будь-який морфологічний варіант (від гіперплазії до аденом) та вражати обидві НЗ. Точна причинна мутація для неї не встановлена, хоча пов’язується з локусом 7p22.
Третя сімейна форма, яка була встановлена нещодавно (2011 р.), є наслідком мутації в гені, що кодує калієві канали KCNJ5 (potassium inwardly rectifying channel, subfamily J, member 5), і також не залежить від впливу АКТГ і лікування глюкокортикоїдами [10].
Не слід забувати також про можливість розвитку третинного гіперальдостеронізму (тобто утворення автономних аденом кори НЗ на тлі тривалої стимуляції секреції альдостерону високим рівнем реніну за реноваскулярної або есенціальної артеріальної гіпертензії), який складно відрізнити від звичайної двобічної ІГК.
Найчастішою формою ПГА є ідіопатична двобічна гіперплазія НЗ (до 70–75 % усіх випадків); аденоми трапляються в 20–30 % випадків. Рідко (< 5 %) спостерігаються також сімейні форми ПГА (глюкокортикоїдчутливі та нечутливі), а також первинна (однобічна) гіперплазія НЗ [2, 5, 6].
Вітчизняний досвід налічує вкрай обмежені клінічні дані, переважно з хірургічних серій спостережень, і тому дослідження структури варіантів ПГА є важливим компонентом вивчення проблеми, на що й була спрямована дана робота.
Матеріали і методи
За період 1996–2014 рр. у клініці та амбулаторії Українського науково-практичного центру ендокринної хірургії, трансплантації ендокринних органів і тканин перебували на лікуванні та обстеженні 474 пацієнти із діагнозом ПГА, із яких 255 хворих було прооперовано в обсязі однобічної адреналектомії, а 219 спочатку отримували консервативне лікування (із них 12 згодом прооперовані).
Пацієнти потрапляли до клініки через випадкове виявлення пухлинних змін у НЗ за даними КТ або МРТ або проходили деталізоване обстеження при пошуку причини резистентної АГ. На перших історичних етапах (1996–2003), ще до становлення систематизованої етапної діагностики ПГА та його конкретних клініко-патогенетичних форм, переважали пацієнти хірургічної групи, які спрямовувались на адреналектомію (відкритим чи лапароскопічним шляхом) після беззаперечної візуалізації поодинокої пухлини НЗ розміром 3–7 см з певними клініко-лабораторними ознаками ПГА (гіперальдостеронемія, гіпокаліємія, тяжка АГ, аритмії). Протягом наступних років (2004–2008 рр.) діагностика ставала більш ретельною, підвищувалась якість рентгенологічних методів візуалізації НЗ, упроваджувались сучасні лабораторні методики, що вплинуло на точність клінічної оцінки захворювання та збільшило частку пацієнтів, які лікувались консервативно. Сучасний етап систематизованого пошуку, підтвердження діагнозу ПГА та встановлення його конкретної клініко-патогенетичної форми сформувався з 2009 року, коли в клініці були впроваджені всі основні визнані міжнародні методи лабораторної та візуалізаційної діагностики.
Так, на сьогодні після скринінгу на ПГА шляхом виділення груп ризику, визначення альдостерон-ренінового співвідношення, а також встановлення можливої гіпокаліємії проводиться обов’язкове підтвердження діагнозу шляхом проведення одного з найбільш надійних діагностичних тестів (навантаження сіллю шляхом внутрішньовенного введення 2,0 л фізіологічного розчину протягом чотирьох годин або каптоприловий тест з прийняттям 25–50 мг каптоприлу та вимірюванням рівня пригнічення альдостерону через 1–2 години) [4]. Для хворих молодого віку та осіб із сімейним анамнезом АГ або початком захворювання з дитинства проводили пробну терапію дексаметазоном для виключення ГКЧА.
Наступним етапом діагностики є візуалізація НЗ. Перевагу віддавали мультиспіральній комп’ютерній томографії (МСКТ) з внутрішньовенним контрастуванням та надтонкими зрізами, хоча подекуди високоінформативною виявлялася й магнітно-резонансна томографія НЗ. За відсутності переконливих даних щодо однобічного ураження НЗ виконували роздільний відбір крові з надниркових вен (РЗКНВ) з вимірюванням рівня альдостерону й кортизолу в надниркових та нижній порожнистій венах.
Пацієнтів, які не бажали лікуватися консервативно та мали переконливі дані щодо однобічного ураження НЗ, спрямовували на лапароскопічну адреналектомію. Видалені препарати НЗ підлягали гістологічному дослідженню з визначенням принципових патологічних змін: дифузна або вузлова гіперплазія, поодинокі чи множинні аденоми (карциноми). На жаль, генетичні методи дослідження й досі недоступні в клінічній практиці та не були застосовані в жодному випадку.
Результати
Одним із завдань дослідження було визначення співвідношення окремих клініко-патогенетичних варіантів ПГА. Однак за об’єктивних причин (відсутність надійних генетичних критеріїв) лише з певним припущенням ми підозрюємо такі форми захворювання, як FH II та FH III, а ГКЧА (FH I) можемо встановити лише на підставі ефективного лікування пацієнтів малими дозами дексаметазону. Особливу форму альдостеронпродукуючої аденоми, яка стимулюється реніном, логічно запідозрити у випадку непригніченого рівня реніну за високого альдостерону та клінічної картини ПГА з однобічною аденомою кори НЗ.
Водночас найбільш важливими клінічними питаннями з точки зору вибору адекватного способу лікування залишаються такі: чи зумовлена секреція альдостерону однобічним ураженням НЗ і пацієнту допоможе хірургічна адреналектомія? Чи може ПГА бути ефективно контрольованим антагоністами мінералокортикоїдних рецепторів або доцільне лікування дексаметазоном (за наявності FH I — ГКЧА)?
Якщо розпочинати з останнього, найменш імовірного варіанта захворювання, то на сучасному рівні медичної генетики було б надзвичайно важливо визначити наявність однієї з відомих мутацій, які спричиняють спадкові (причому завжди автосомно-домінантні) варіанти ПГА, що охоплюють 2–5 % усіх випадків хвороби. Наразі таке дослідження в Україні вбачається нереальним навіть з науковою метою, не кажучи про широке практичне використання. Тому в розпорядженні науковців та лікарів залишається клініко-генеалогічний метод, що базується на ретельному зборі анамнезу, дослідженні кровних родичів пацієнта, визначенні низки гормональних параметрів та проведенні пробного лікування. Останнє стосується насамперед FH I — ГКЧА, коли завдяки химеризації генів, що відповідають за синтез альдостерону та рецепторів до АКТГ, патологічна гіперсекреція альдостерону стимулюється навіть нормальними концентраціями кортикотропіну крові. Для цієї форми дуже ефективним є спосіб лікування малими дозами синтетичних глюкокортикоїдів (дексаметазон, преднізолон), які за принципом зворотного зв’язку пригнічують рівень АКТГ та далі — секрецію альдостерону.
Для цього варіанта притаманним є ранній початок хвороби (у юнацькому або навіть дитячому віці) з підвищенням АТ до помірних та високих цифр, наявність кровних родичів зі схожою клінікою, частими цереброваскулярними ускладненнями АГ (причому не стільки через підвищення тиску та звуження судин, як через генетично детерміновані аномалії судинної стінки з мікроаневризмами та телеангіектазіями).
З огляду на особливості патогенезу цієї форми ПГА та враховуючи обмежені можливості генетичної діагностики, ми запропонували після ретельного вивчення анамнезу захворювання та обстеження родичів пацієнта першої лінії, а також після виключення поодинокої пухлини НЗ застосувати для пацієнтів із підозрілими клініко-анамнестичними даними пробне лікування малими дозами дексаметазону (0,5–1,0 мг один раз ввечері протягом 4–7 діб). Рівень альдостерону, калію та натрію крові перевіряли до та після лікування, а показники АТ моніторували постійно. Виняток робили для пацієнтів із непригніченим кортизолом крові після нічної дексаметазонової проби, пацієнтів із підвищеним рівнем АКТГ, із пухлинами в НЗ або з віком початку захворювання (підвищення АТ) понад 40 років.
Після згаданих виключень підозра на можливість існування глюкокортикоїдчутливої форми спадкового ПГА залишилась дійсною для 22 пацієнтів із 219 хворих групи консервативного лікування. Пробне лікування підтвердило діагноз у чотирьох пацієнтів (1,8 %), які з успіхом продовжили лікування зі зменшенням дози дексаметазону до мінімальної (0,25–0,5 мг на добу), яка має достатній клінічний ефект (нормалізація рівня альдостерону крові, АТ та підтримання нормокаліємії). Ще в двох пацієнтів із двобічними гіперпластичними змінами в НЗ була запідозрена наявність іншої сімейної форми ПГА — FH II, враховуючи спадковий характер хвороби.
Для решти 213 пацієнтів необхідним був наступний етап диференціальної діагностики між ідіопатичною двобічною гіперплазією надниркових залоз та первинною (однобічною) гіперплазією кори надниркової залози.
МСКТ із внутрішньовенним контрастуванням на сучасних апаратах із зрізами завтовшки 1–2 мм може надати дуже корисну та детальну інформацію щодо структурних змін у НЗ у пацієнтів із ПГА. На жаль, ця інформація не несе відомостей щодо функціональної активності НЗ та не може у великому відсотку випадків дати відповідь на питання, чи одна надниркова залоза виділяє надлишок альдостерону, чи гіперсекреція спричинена обома НЗ. На це питання з найбільшою об’єктивністю може відповісти РЗКНВ. Водночас ця процедура є найбільш відповідальною, потребує високих професійних навичок та досвіду виконавців, несе з собою певні ризики та потребує відповідного фінансового забезпечення через високу вартість необхідних витратних матеріалів та реактивів. Тому багато пацієнтів або відмовлялися від проведення РЗКНВ за різних причин, або не могли її отримати через певні обставини (ожиріння, гіпокоагуляція, супутня гіперкортизолемія).
Усього виконано 63 процедури РЗКНВ. Для 14 з них (22,2 %) було зроблено висновок щодо переважно однобічної гіперсекреції альдостерону та запропоновано хірургічне лікування. У подальшому операцію виконано в 12 пацієнтів із позитивним функціональним результатом та гістологічним підтвердженням дифузної або вузликової гіперплазії НЗ (9) та аденом НЗ (у двох випадках). В одному випадку ефекту від адреналектомії не спостерігалось, що вказувало на двобічний характер ураження НЗ і відповідало варіанту ІГК.
Крім цих 12 прооперованих за даними РЗКНВ пацієнтів ще 255 хворим хірургічне лікування було призначене відразу за даними візуалізаційного обстеження з доведеним чи запідозреним ПГА (без проведення РЗКНВ). Аргументом на користь лапароскопічної адреналектомії слугували наявність поодинокої пухлини НЗ розміром 0,7–9,5 см за даними МСКТ, підозра на злоякісність, ріст пухлини протягом 6–12 міс. спостереження, бажання хворого. Гістологічне дослідження показало наявність поодинокої кортикальної аденоми НЗ у 193 хворих, що цілковито підтверджувало початковий діагноз АПА (синдром Конна) та відповідало клінічному перебігу та позитивним результатам лікування. У двох пацієнтів підтверджено злоякісний характер пухлини. Один із цих хворих з адренокортикальним раком помер через інвазивний та метастатичний характер онкологічного процесу. Для решти 60 пацієнтів був отриманий гістологічний висновок: вузликова гіперплазія кори НЗ. Відмінний результат лікування зафіксовано тільки для 16 пацієнтів із цієї групи (свідчить на користь ПГНЗ), тоді як у решти 44 хворих зафіксовано лише певне полегшення перебігу АГ та зниження рівня альдостерону. Для 35 з цих 44 пацієнтів згодом було встановлено рецидив ПГА, що вказує на ймовірну двобічну гіперплазію НЗ (ІГП) та можливу ПГНЗ у решти 9.
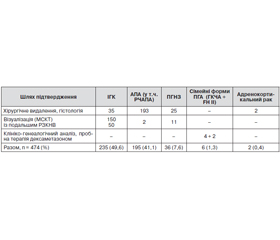
Отже, весь масив із 474 пацієнтів, які перебували на лікуванні з діагнозом ПГА, можна з певними припущеннями розділити на такі основні групи: двобічна ідіопатична гіперплазія НЗ — 235, альдостеронпродукуюча аденома (в тому числі РЧАПА) — 195, первинна (однобічна) гіперплазія НЗ — 36, сімейні форми — 6 (у тому числі ГКЧА — 4, FH II — 2), адренокортикальний рак — 2.
Обговорення
Загальний розподіл клініко-патогенетичних варіантів ПГА та діагностичних шляхів їх виявлення наведено в табл. 1.
/135/135.jpg)
Як видно з наведених даних, основною формою ПГА в серії клінічних спостережень, накопичених за період 1996–2014 років у спеціалізованому ендокринологічному науковому закладі, є двобічна ідіопатична гіперплазія НЗ, яка може мати характер як дифузної, так вузлової гіперплазії й становить близько половини всіх випадків. Це збігається з літературними даними, але дещо нижче за рахунок серії хворих, які лікувались переважно хірургічно внаслідок виявлення пухлини НЗ, що збільшило відносну частоту аденом. Другою за частотою формою ПГА, яка в загальній кількості хворих спостерігається дедалі рідше, є поодинока альдостеронпродукуюча аденома, відома як синдром Конна. Її частка становить 41 %. Ми не змогли визначити дійсну кількість випадків ренінзалежних аденом (РЧАПА) через відсутність динамічного визначення реніну в крові в пацієнтів, а також недостатню кількість інформації в патогістологічних висновках щодо походження епітелію пухлин (із сітчастої або клубочкової зони кори НЗ). Адже для АПА характерним є походження з клітин сітчастої зони, а для РЧАПА та ПГНЗ — із клубочкової. Менш часто (7,6 %) спостерігається первинна гіперплазія НЗ, яку слід шукати перш за все за непевних візуалізаційних даних обстеження пацієнтів із ПГА за допомогою МСКТ. Надійним методом підтвердження цієї форми може бути лише РЗКНВ, що має зайняти чільне місце в арсеналі клінічної діагностики ПГА в спеціалізованих закладах.
Частка спадкових форм є невеликою (менше 2 %), водночас можливість надійного лікування ГКЧА малими дозами дексаметазону потребує застосування пробної терапії у випадках підозри за даними клініко-генеалогічного дослідження пацієнтів, насамперед молодого віку. Генетичні дослідження в майбутньому дадуть змогу об’єктивно підтверджувати спадковий варіант ПГА.
Висновки
1. Проведені дослідження підтверджують відомості щодо найбільшого поширення двобічної ідіопатичної гіперплазії НЗ як переважної клініко-патогенетичної форми ПГА.
2. Частка альдостеронпродукуючих аденом НЗ має тенденцію до зменшення в загальному масиві причин ПГА та становить загалом близько 40 %.
3. Для виявлення більш рідкісної форми ПГА — первинної гіперплазії НЗ необхідне проведення РЗКНВ.
4. Розвиток медичної генетики дозволить у майбутньому об’єктивізувати діагностику сімейних форм ПГА, які зараз підтверджуються лише пробною терапією дексаметазоном та клініко-генеалогічним дослідженням.
1. Comparison of active renin concentration and plasma renin activity for the diagnosis of primary hyperaldosteronism in patients with an adrenal mass [Text] / N. Unger, I.L. Schmidt, C. Pitt [et al.] // Eur. J. Endocrinol. — 2004. — Vol. 150. — P. 517–523.
2. Diagnosis and treatment of primary hyperaldosteronism [Text] / J.D. Blumenfeld, J.E. Sealey, Y. Schlussel [et al.] // Ann. Intern. Med. — 1994. — Vol. 121. — P. 877–885.
3. Fardella C.E. Primary hyperaldosteronism in essential hypertensives: prevalence, biochemical profile, and molecular biology [Text] / C.E. Fardella, L. Mosso, C.E. Gomez-Sanchez [et al.] // J. Clin. Endocrinol. Metab. — 2000. — Vol. 85. — P.1863–1867.
4. Funder J.W. Case Detection, Diagnosis, and Treatment of Patients with Primary Aldosteronism: An Endocrine Society Clinical Practice Guideline [Text] / J.W. Funder, R.M. Carey, C. Fardella, C.E. Gomez-Sanchez, F. Mantero, M. Stowasser, W.F.Jr. Young, V.F. Montori // J. Clin. Endocr. Metab. — 2008. — Vol. 93, № 9. — P. 3266–3281.
5. Ganguly A. Primary Aldosteronism [Text] / A. Ganguly // N. Engl. J. Med. — 1998. — Vol. 339, № 25. — P. 1828–1834.
6. Halperin F. Glucocorticoid-remediable aldosteronism [Text] / F. Halperin, R.G. Dluhy // Endocrinol. Metab. Clin. North. Am. — 2011. — Vol. 40, № 2. — P. 333–341.
7. Montori V.M. Use of plasma aldosterone concentration to plasma renin activity ratio as a screening test for primary aldosteronism. A systematic review of the literature [Text] / V.M. Montori, W.F. Young Jr. // Endocrinol. Metab. Clin. North Am. — 2002. — Vol. 31. — P. 619–632.
8. Rossi G.P. Surgically correctable hypertension caused by primary aldosteronism // Best Pract. Res. Clin. Endocrinol. Metab. — 2006. — Vol. 20. — P. 385–400.
9. Schwarz G.L. Screening for primary aldosteronism in essential hypertension: diagnostic accuracy of the ratio of plasma aldosterone concentration to plasma renin activity [Text] / G.L. Schwarz, S.T. Turner // Clin. Chem. — 2005. — Vol. 51. — P. 386–394.
10. Choi M. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension [Text] / Choi M., U.I. Scholl, P. Yue [et al.] // Science. — 2011. — Vol. 331 (6018). — Р. 768–772.