
Международный эндокринологический журнал 2 (66) 2015
Вернуться к номеру
Орфанні спадкові синдроми у практиці педіатра-ендокринолога
Авторы: Ризничук М.О., Пішак В.П. — Буковинський державний медичний університет, м. Чернівці
Рубрики: Эндокринология
Разделы: Справочник специалиста
Версия для печати
У лекції подано коротку фенотипово-генетичну характеристику орфанних ендокринних синдромів, що супроводжуються гігантизмом.
В лекции дана краткая фенотипически-генетическая характеристика орфанных эндокринных синдромов, сопровождающихся гигантизмом.
The lecture presents short phenotypic and genetic characteristics of orphan endocrine syndromes associated with gigantism.
орфанні спадкові синдроми, гігантизм, діти, гени.
орфанные наследственные синдромы, гигантизм, дети, гены.
orphan hereditary syndromes, gigantism, children, genes.
Статья опубликована на с. 153-157
Ендокринна патологія спадкового генезу набуває значного поширення. Серед хвороб, що трапляються досить часто, виділяють цукровий діабет 1-го типу, ураження щитоподібної та надниркових залоз. Успіхи молекулярної біології сприяли розкриттю патогенезу рідкісних ендокринних хвороб із суттєвим генетичним компонентом.
Рідкісні (орфанні) захворювання — це такі, що трапляються з певною частотою, набувають хронічного прогресуючого прогредієнтного перебігу, а без лікування загрожують життю чи спричиняють пожиттєву інвалідизацію пацієнтів.
До орфанних належать захворювання (синдроми), поширеність яких становить 10 випадків на 100 000 населення. Серед численної групи таких хвороб, а їх понад 7000, прискіпливої уваги потребують хвороби ендокринного генезу із суттєвими генетичними відхиленнями. Фенотипові ознаки таких захворювань або виявляють з народження, або вони виникають у дитячому віці.
Розглянемо спадкові синдроми, у клінічному перебігу яких переважають явища гігантизму в поєднанні з ендокринною патологією.
Перлмана синдром (MIM 267000)
Синоніми: гамартома нирки, нефробластома та гігантизм плода; фетальний асцит, макросомія плода та пухлина Вільмса.
Вперше описаний Liban і Kozenitzky (1970) [13]. Perlman et al. (1975) описали 5 випадків єврейсько-єменської сім’ї, які були двоюрідними сибсами, з вадами, що проявлялися макросомією при народженні, двобічними нирковими гамартомами з нефробластоматозом або без нього, гіпертрофією острівців Лангерганса і незвичайним обличчям [17].
До фенотипових ознак належать асцит без набряку плода і багатоводдя [18].
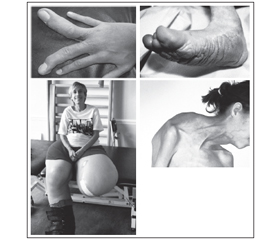
Клінічні прояви. Хворі діти великої маси при народженні, гіпотонія м’язів, органомегалія, характерний лицевий дизморфізм (перевернута V-подібна верхня губа, відкритий рот, опуклий лоб, глибоко посаджені очі, широка і плоска спинка носа, низько посаджені вуха), ниркові аномалії (нефромегалія і гідронефроз), агенезія мозолистого тіла, затримка психомоторного розвитку, гіперінсулінізм і висока смертність новонароджених (рис. 1).
/154/154.jpg)
Синдром Перлмана зумовлює високий ризик виникнення пухлини Вільмса. З 64 % випадків у новонароджених, які вижили в неонатальному періоді, пухлину діагностували в більш ранньому віці порівняно зі спорадичними випадками (менше 2 років і 3–4 роки відповідно). Доведена висока частота двобічних пухлин (55 %). Гістологічне дослідження нирок у дітей із синдромом Перлмана виявляє частий нефробластоматоз, що передує пухлині Вільмса.
Синдром Перлмана зумовлений гомозиготною або гетерозиготною мутацією в гені DIS3L2 (МІМ 614184) на хромосомі 2q37 [11].
Тип успадкування — автосомно-рецесивний.
Протея синдром (МІМ 176920)
Синоніми: гігантизм, частковий, руки і ноги, NEVI, гемігіпертрофія, І макроцефалія, людина-слон.
Вперше описаний M. Cohen і P. Hayden у 1979 р., які і дали назву на честь древньогрецького бога Протея (грец. Proteus — «поліморфний»), який мав здатність змінювати форму свого тіла з метою більш успішного захоплення здобичі [6].
Причиною синдрому є спонтанна варіація гена під час ембріонального розвитку. Тяжкість захворювання залежить від періоду ембріогенезу, коли власне виникла генетична мутація, що призводить щонайменше до утворення трьох підвидів клітин — нормальних, атрофічних і гіпертрофічних [7, 12].
У хворих на синдром Протея підвищена активність білка AKT, у здорових людей цей білок неактивний. Мутація проявляє себе як прискорювач росту клітин, але не у всіх тканинах тіла [14, 21].
Всього у світі налічується близько 120 осіб із цим невиліковним захворюванням [22].
Клінічні прояви. Синдром Протея супроводжується розростанням шкіри, кісток, м’язів, жирової тканини, кровоносних і лімфатичних судин. При народженні діти, як правило, не мають очевидних ознак захворювання. Фенотипові прояви синдрому Протея виявляють у перші 2 роки життя дитини [23, 24].
Діагностується косоокість, екзофтальм, міопія, прогенія, варикозне розширення вен, розростання шкіри на підошвах (рис. 2).
/155/155.jpg)
Приблизно у 55 % випадків виявляється розумова відсталість, у 13 % — судомний синдром. Траплялися також хворі з ізольованою макродактилією, спленомегалією, вибірковою патологією очей і черепа у вигляді множинних менінгіом, полімікрогірією, ретинальною пігментною дегенерацією та атрофією зорового нерва. Тривалість життя пробандів, як правило, перебуває в межах від 3 до 40 років [10, 25].
З віком спостерігається надмірно швидкий ріст пухлин. Середня тривалість життя у хворих часто скорочується від підвищення тромбозу глибоких вен та емболії легеневої артерії як ускладнення хвороби судинною мальформацією.
Розростання тканин може призвести до вторинного пошкодження нервової системи, що, у свою чергу, спричиняє когнітивні порушення.
Хворим загрожує підвищений ризик розвитку певних видів раку, менінгіоми й аденоми слинних залоз.
Синдром пов’язаний із мутацією в гені AKT1 (МІМ 164730) на хромосомі 14q32.3.
Мутації AKT1 модифікують активність клітинного білка AKT, що стимулює ріст.
Тип успадкування — автосомно-домінантний.
Сотоса 1 синдром (МІМ 117550)
Синонім: SOTOS 1 синдром, мозковий гігантизм, делеції хромосоми 5q35 синдром.
Синдром Сотоса, або синдром церебрального гігантизму, вперше описаний американським педіатром J. Sotos у 1964 р. [5].
Він виникає з частотою 1 : 10 000–50 000 новонароджених. У світовій літературі описано близько 120 випадків. Серед хворих переважають хлопчики.
Оскільки більшість описаних випадків носить спорадичний характер, раніше багато з них розцінювалися як приклад автосомно-рецесивного успадкування синдрому Сотоса. Проте зараз ці дані переглядаються і висувається припущення, що це неповна пенетрантність патологічного гена, гонадного мозаїцизму або генетичної гетерогенності хвороби [3].
Клінічні прояви. Синдром надлишкового росту дітей, що характеризується надмірним зростом, певними краніолицевими ознаками, затримкою психічного розвитку та збільшенням кісткового віку.
За відсутності одного або більше з чотирьох критеріїв (ріст більше 97-го процентиля, окружність голови більше 97-го процентиля, кістковий вік більше 90-го процентиля, затримка розумового розвитку) діагноз малоймовірний.
Захворювання маніфестує з народження. Привертають увагу великі розміри новонародженої дитини. Середня довжина тіла становить 55 см, маса — 3900 г, на 3–5-му роках життя спостерігається інтенсивне збільшення росту. Типові й черепно-лицьові дизморфії: макродоліхоцефалія, виступаючі лобні бугри, грубі риси обличчя з гіпертелоризмом, страбізм, антимонголоїдний розріз очей, виступаюча нижня щелепа (прогнатизм), макроглосія й готичне піднебіння. Кісткові зміни проявляються такими особливостями: великі кисті і стопи, кіфосколіоз, синдактилія стоп. Зі сторони внутрішніх органів іноді виявляється вісцеромегалія. Можливі вади розвитку серця, нирок, центральної нервової системи. В окремих випадках бувають судоми і порушення координації [5]. Ступінь розумової відсталості зазнає певних відхилень, але зазвичай буває помірним. Кінцевий ріст знаходиться на верхньому перцентилі. Зріст дорослих зазвичай не виходить за межі нормальних значень, у чоловіків він становить 184,3 ± 6,0 см, у жінок 172,9 ± 5,7 см.
Спостерігаються аномальна дерматогліфіка, нормальний рівень гормона росту і високий рівень валіну, ізолейцину та лейцину в крові.
Рентгенофункціональні методи дослідження виявляють випередження кісткового віку, неспецифічні зміни на ЕКГ, розширення шлуночків мозку. На МРТ нерідко знаходять гіпоплазію мозолистого тіла. У дитинстві випередження кісткового віку становить 2–4 роки, статеве дозрівання може настати відносно рано, хоча залишається в межах нормального діапазону.
Дана патологія іноді поєднується з розвитком злоякісних новоутворень (пухлина Вільмса, рак печінки, яєчників) [2].
Викликається гетерозиготною мутацією в гені NSD1 (МІМ 606681) у ділянці 5q35 [16].
Тип успадкування — автосомно-домінантний, більшість випадків спорадичні. Тривалість життя не змінена. Прогноз хороший, лікування не проводять.
Сотоса 2 синдром (МІМ 614753)
Синоніми: SOTOS 2 синдром, мозковий гігантизм, делеції хромосоми 5q35 синдром.
Викликається гетерозиготною мутацією в гені NFIX (МІМ 164005) на хромосомі 19p13.3 [8].
Malan et al. (2010) повідомили про 3 незв’язаних випадки з подібним фенотипом, а саме: інтенсивний післяпологовий лінійний ріст, макроцефалія, випередження кісткового віку, довге вузьке обличчя, високе чоло, сколіоз. Незвичайна поведінка характеризується розвитком тривожності та розумової відсталості.
Берардінеллі синдром (МІМ 269700)
Синоніми: синдром Берардінеллі — Сейпа, синдром Брунзелла, ліпоатрофічний діабет, уроджена генералізована ліподистрофія, BSCL.
Синдром Берардінеллі — Сейпа описаний аргентинським ендокринологом W. Berardinelli та норвезьким педіатром M. Seipе [4].
Це автосомно-рецесивне захворювання, що трапляється з частотою 1 : 10 000 000 осіб та характеризується практично повною відсутністю підшкірної клітковини [15, 19].
Молекулярно-генетичним аналізом доведено існування чотирьох типів уродженої генералізованої ліподистрофії [9, 20].
Тип 1 зумовлений мутаціями в гені AGPAT2, що кодує 1-ацил-гліцерол-О-ацетилтрансферазу 2. Ген розташований у локусі 9q34. Продукт гена білок AGPAT2 складається з 278 амінокислот і забезпечує біосинтез тригліцеридів і гліцерофосфоліпідів. Зниження рівня білка AGPAT2 у жировій тканині призводить до ліподистрофії внаслідок порушення синтезу тригліцеридів чи патологічної функції адипоцитів (відсутні фосфоліпіди) [2].
И.И. Дедов, М.В. Шестакова [1] підтерджують ліпоатрофічний цукровий діабет як рідкісне захворювання вродженого чи набутого характеру. Симптоматика генералізованої атрофії підшкірної жирової клітковини на тлі вираженої інсулінорезистентності й гіперінсулінемії в поєднанні з гіпертрофією скелетних м’язів, гіпертригліцеридемією й артеріальною гіпертензією. Автори акцентують увагу, що захворювання викликане генетичними дефектами адипогенезу і/або прискореним апоптозом адипоцитів, порушеною секрецією адипоцитокінів (лептину, адипонектину), що забезпечують метаболізм жирової тканини.
У лютому 2014 р. вперше зареєстрований препарат для лікування цього синдрому — рекомбінантний аналог лептину (метрелептин) [15].
Тип 2 синдрому розвивається внаслідок мутацій у гені BSCа2 в локусі 11q13. Продукт гена білок сейпін BSCа2 містить 398 амінокислот, причетний до диференціювання та функціонування адипоцитів.
Молекулярно-генетичну основу синдрому Берардінеллі — Сейпа типу 3 становлять мутації в гені CAV1 (локус 7q31), за участі якого синтезується білок кавеолін 1 — невід’ємна складова мембранних везикул. Зазначений білок локалізується на мембранах адипоцитів і зв’язує жирні кислоти, забезпечує транспорт ліпідів.
Уроджена генералізована ліподистрофія типу 4 спричинена мутаціями в гені PTRF, розташованому на хромосомі 17, і кодує білок PTRF (polymerase and transcript release factor). Мутації в зазначеному гені зумовлюють повну втрату функції відповідного білка, порушується локалізація кавеолінів у скелетних м’язах.
Клінічні прояви. Перші симптоми вродженої генералізованої ліподистрофії виявляють від народження або на першому році життя. Основними лабораторними ознаками захворювання є тяжка гіпертригліцеридемія, низька концентрація ліпопротеїнів високої щільності у крові, інсулінорезистентність, значне зниження рівня лептину та адипонектину. У підлітковому віці інсулінорезистентність призводить до розвитку цукрового діабету. Відбувається аномальне накопичення жиру в різних органах (печінка, м’язи, серце), що спричиняє розвиток тяжких клінічних ускладнень: ожиріння печінки, цирозу, нефропатії, гіпертрофічної кардіоміопатії, серцевої недостатності та миттєвої смерті.
Хворим властивий швидкий ріст, підвищений апетит та особлива зовнішність. Відсутність підшкірної жирової тканини в поєднанні зі збільшенням м’язової тканини формує в таких хворих виражений м’язовий рельєф. Інші ознаки: велике підборіддя, великі кисті рук та стоп, гірсутизм, acantosis nigrigans. Хворим жінкам властивий збільшений клітор, а в чоловіків збільшуються зовнішні статеві органи.
У більшості випадків ознаки цукрового діабету розвиваються в другій декаді життя хворої людини. Ускладненнями синдрому є гіпертрофічна кардіоміопатія, жирова дистрофія печінки, м’язова гіпертрофія. Різні ознаки свідчать про патологію ендокринної системи (швидкий ріст, передчасне статеве дозрівання та інші), часто спостерігається утворення кіст кісток, з якими пов’язують часті переломи.
Клінічний діагноз виставляють за наявності трьох основних критеріїв або двох основних плюс два або більше малі критерії.
Основні критерії:
— ліпоатрофія тулуба, кінцівок і обличчя. Загальна ліподистрофія з’являється при народженні. У деяких осіб ліподистрофія виникає впродовж перших місяців життя. Ліпоатрофія зумовлює спортивну зовнішність, тому що скелетні м’язи гіпертрофовані;
— акромегалоїдні ознаки — гігантизм, м’язова гіпертрофія, випередження кісткового віку, прогнатизм, збільшені руки і ноги, кліторомегалія і збільшені зовнішні статеві органи в чоловіків;
— гепатомегалія;
— підвищені концентрації в сироватці крові тригліцеридів до 80 г/л, іноді асоціюються з гіперхолестеринемією;
— інсулінорезистентність. Підвищені концентрації в сироватці крові інсуліну і С-пептиду можуть спостерігатися з перших років життя. Виражені клінічні прояви цукрового діабету зазвичай розвиваються у другому десятилітті. Рання клінічна ознака — чорний акантоз шиї і пахвових западин, що в окремих випадках набувають бородавчастого вигляду.
Малі критерії:
— гіпертрофічна кардіоміопатія може з’являтися в дитинстві або розвиватися пізніше;
— психомоторні помірні (IQ 50–70) або глибокі (IQ 35–50) інтелектуальні порушення. Близько 80 % осіб із мутаціями в гені BSCL2 властиві легкі або помірні інтелектуальні порушення, у той час як тільки 10 % осіб із мутаціями в гені AGPAT2 мають тяжкі інтелектуальні порушення;
— гірсутизм проявляється низьким ростом волося на чолі та шиї; гіпертрихоз, мабуть, не залежить від гормональної стимуляції;
— передчасне статеве дозрівання у жінок (у 10 %);
— кісти кісток трапляються у 8–20 % хворих осіб і мають полікістозні прояви на рентгенограмі. Розташовані в епіфізарних і метафізарних ділянках довгих трубчастих кісток. Кісти кісток часто діагностуються в другій декаді життя і в основному спостерігаються в осіб із мутаціями в гені AGPAT2;
— мегалія м’язів нижніх і верхніх кінцівок відносна і спостерігається, зокрема, через відсутність підшкірного жиру.
Цим повідомленням ми зробили спробу привернути увагу лікарів-практиків до нечітко окресленої групи орфанних захворювань. Вочевидь, назріла необхідність єдиного підходу до їх визначення, класифікації, критеріїв діагностики. Такі кроки доцільні ще й тому, що найближчим часом очікується черговий перегляд Міжнародної статистичної класифікації хвороб та проблем, пов’язаних зі здоров’ям, 10-го перегляду (МКХ-10).
1. Дедов И.И. Персонализированная терапия сахарного диабета: путь от болезни к больному / И.И. Дедов, М.В. Шестакова // Тер.архив. — 2014. — № 10. — С. 4–9.
2. AGPAT2 is mutated in congenital generalized lipodystrophy linked to chromosome 9q34 / A.K. Agarwal, E. Arioglu, S. de Almeida [et al.] // Nature Genet. — 2002. — V. 31. — P. 21–23.
3. Apparent Sotos syndrome (cerebral gigantism) in a child with Trisomy 20p11.2-p12.1 mosaicism / L. Faivre, G. Viot, M. Prieur [et al.] // Am. J. Med. Genet. — 2000. — V. 91. — P. 273–276.
4. Berardinelli W. An undiagnosed endocrino-metabolic syndrome: report of two cases / W. Berardinelli // The J. of Clin. Endocrinol. and Metabol. — 1954. — V. 14. — P. 193–204.
5. Cerebral gigantism in childhood. A syndrome of excessively rapid growth with acromegalic features and a nonprogressive neurologic disorder / J.F. Sotos, P.R. Dodge, D. Muirhead [et al.] // New England J. of Medicine, Boston. — 1964. — V. 271. — P. 109–116.
6. Cohen M.M. Jr. A new recognized hamartomatous syndrome / M.M. Cohen Jr., P.W. Hayden // Birth. Defects Orig. Art. — 1979. — V. 15, № 5B. — P. 291–296.
7. Cohen M.M. Jr. Proteus syndrome: clinical evidence for somatic mosaicism and selective review / M.M. Cohen Jr. // Am. J. Med. Genet. — 1993. — V. 47. — P. 645–652.
8. Deutliche Auswirkungen der allelischen NFIX Mutationen auf Nonsense-vermittelten mRNA-Zerfall erzeugen entweder ein Sotos-like oder ein Marshall-Smith-Syndrom / V. Malan, D. Rajan, S. Thomas [et al.] // Am. J. Hum. Genet. — 2010. — V. 87. — P. 189–198.
9. Gene and phenotype analysis of congenital generalized lipodystrophy in Japanese: a novel homozygous nonsense mutation in seipin gene / K. Ebihara, T. Kusakabe, H. Masuzaki [et al.] // J. Clin. Endocr. Metab. — 2004. — V. 89. — P. 2360–2364.
10. Germline and germline mosaic PTEN mutations associated with a Proteus-like syndrome of hemihypertrophy, lower limb asymmetry, arteriovenous malformations and lipomatosis / X.-P. Zhou, D.J. Marsh, H. Hampel [et al.] // Hum. Mol. Genet. — 2000. — V. 9. — P. 765–768.
11. Germline mutations in DIS3L2 cause the Perlman syndrome of overgrowth and Wilms tumor susceptibility / D. Stuti, M.R. Morris, W.N. Cooper [et al.] // Nature Genet. — 2012. — V. 44. — P. 277–284.
12. Kruger G. Transmission of Proteus syndrome from mother to son? (Letter) / G. Kruger, L. Pelz, H.-R. Wiedemann // Am. J. Med. Genet. — 1993. — V. 45. — P. 117–118.
13. Liban E. Metanephric hamartomas and nephroblastomatosis in siblings / E. Liban, I.L. Kozenitzky // Cancer. — 1970. — V. 25. — P. 885–888.
14. Mohamedbhai A.G. Neonatal Proteus syndrome? (Letter) / A.G. Mohamedbhai, A.M.H. Miyan, D. Lacombe // Am. J. Med. Genet. — 2002. — V. 112. — P. 228–230.
15. Nainggolan L. Myalept (metreleptin) Approved for Generalized Lipodystrophy / L. Nainggolan // Medscape Medical News. February 25, 2014. Available at: http://www.medscape.com/viewarticle/82105625, 2014.
16. NSD1 mutations are the major cause of Sotos syndrome and occur in some cases of Weaver syndrome but are rare in other overgrowth phenotypes / J. Douglas, S. Hanks, I.K. Temple [et al.] // Am. J. Hum. Genet. — 2003. — V. 72. — P. 132–143.
17. Perlman M. Syndrome of fetal gigantism, renal hamartomas and nephroblastomatosis with Wilms’ tumour / M. Perlman, M. Levin, B. Wittels // Cancer. — 1975. — V. 35. — P. 1212–1217.
18. Perlman syndrome: report, prenatal findings and review / J.-L. Alessandri, F. Cuillier, D. Ramful [et al.] // Am. J. Med. Genet. — 2008. — V. 146A. — P. 2532–2537.
19. Seip M. Lipodystrophy and gigantism with associated endocrine manifestation: a new diencephalic syndrome? / M. Seip // Acta paediatrica. — 1959. — V. 48. — P. 555–574.
20. Severe cardiac phenotype of Berardinelli-Seip congenital lipodystrophy in an infant with homozygous E189X BSCL2 mutation / B. Friguls, W. Coroleu, R. del Alcazar [et al.] // Europ. J. Med. Genet. — 2009. — V. 52. — P. 278–279.
21. Smeets E. Regional Proteus syndrome and somatic mosaicism / E. Smeets, J.-P. Fryns, M.M. Cohen Jr. // Am. J. Med. Genet. — 1994. — V. 51. — P. 29–31.
22. Sudden death caused by pulmonary thromboembolism in Proteus Labell syndrome / A.M. Slavotinek, S.J. Vacha, K.F. Peters [et al.] // Clin. Genet. — 2000. — V. 58. — P. 386–389.
23. The Proteus syndrome: partial gigantism of the hands and/or feet, nevi, hemihypertrophy, subcutaneous tumors, macrocephaly or other skull anomalies and possible accelerated growth and visceral affections / H.-R. Wiedemann, G.R. Burgio, P. Aldenhoff [et al.] // Eur. J. Pediat. — 1983. — V. 140. — P. 5–12.
24. Transmission of Proteus syndrome from father to son? / J. Goodship, A. Redfearn, D. Milligan [et al.] // J. Med. Genet. — 1991. — V. 28. — P. 781–785.
25. Waite K.A. Protean PTEN: Form and Function / K.A. Waite, C. Eng // Am. J. Hum. Genet. — 2002. — V. 70. — P. 829–844.