Статья опубликована на с. 111-117
Гиалиноз (hyalinosis; греч. hyalinos — прозрачный, стекловидный; синонимы: гиалиновое перерождение, гиалиновая дистрофия) — один из видов белковой внеклеточной дистрофии, при которой в ткани образуются однородные полупрозрачные плотные массы, напоминающие гиалиновый хрящ. Гиалиноз может явиться проявлением общих нарушений белкового обмена, однако чаще всего это местный очаговый или же системный дистрофический процесс. Понятие «гиалиноз» объединяет различные по происхождению, механизму развития и биологической сущности процессы. Основное в развитии гиалиноза — деструкция волокнистых структур соединительной ткани и повышение тканево-сосудистой проницаемости в связи с ангионевротическими (дисциркуляторными), метаболическими, воспалительными и иммунопатологическими процессами. В результате нарушения проницаемости происходит пропитывание ткани плазменными белками и адсорбция их неизмененных волокнистых структур с последующей преципитацией. Образующийся гиалин имеет различный, в зависимости от характера заболевания, химический состав (например, гиалин при диабетической микроангиопатии и гиалин при так называемых иммунокомплексных заболеваниях). В большинстве случаев процесс необратим, реже возможно и рассасывание гиалиновых масс. В некоторых случаях гиалиноз может рассматриваться как физиологический процесс, например гиалиноз сосудов селезенки людей зрелого и пожилого возраста.
Синдром гиалинового фиброматоза является редким, фатально прогрессирующим, аутосомно-рецессивным заболеванием со смертельным исходом, характеризующимся выраженными контрактурами суставов, множественными подкожными узелками, гипертрофией десен, остеопенией, задержкой психомоторного и речевого развития. Это обусловлено мутациями в ANTXR2 (также известный как CMG2) гене, который кодирует трансмембранный белок, участвующий в образовании базальной мембраны эндотелия и сборке внеклеточного матрикса. Мутация этого гена приводит к нарушению формирования базальной мембраны, что позволяет гиалиновому веществу накапливаться в различных тканях организма, что и проявляется развитием подкожных узелков, контрактур суставов, остеолизом [9, 13, 19, 21, 22].
Выделяют несколько вариантов синдрома гиалинового фиброматоза:
— инфантильный системный гиалиноз (infantile systemic hyalinosis, ISH) — прогностически неблагоприятный вариант, при котором в большинстве случаев дети умирают в первые годы жизни;
— ювенильный гиалиновый фиброматоз (juvenile hyalinic fibromatosis, JHF), представляющий более мягкие формы заболевания, первоначально описанные у подростков.
Инфантильный системный гиалиноз и ювенильный гиалиновый фиброматоз — два варианта одного и того же заболевания, в настоящее время совместно называются синдромом гиалинового фиброматоза (hyalinic fibromatosis syndrome, HFS). Этот термин был впервые введен Нофал и др. [16, 22]. Первый медицинский случай HFS описан в 1873 году в одной английской семье. Болезнь сначала была названа фиброзирующим моллюском [15, 21, 23], а затем в 1962 году Puretici и др. сообщили о случае под названием «мезенхимальная дисплазия». В 1964 году Исикава и Хори ввели термин «системный гиалиновый фиброматоз» [14].
JHF является редким, наследуемым по аутосомно-рецессивному типу заболеванием, с различными клиническими и гистологическими особенностями. Обзор литературы выявил более 60 случаев, зарегистрированных во всем мире [15]. Манифестация симптоматики отмечается в течение первых двух лет жизни, а в дальнейшем быстро прогрессирует. Гипертрофия десен чаще развивается в течение первого года жизни. Элементы сыпи (узелки и папулы) локализуются больше в области ушей и носа, половых органов и на бедрах. Очень часто кожные повреждения подвергаются инфицированию. Развиваются сочетанные, прогрессирующие и болезненные контрактуры суставов, из-за которых пациенты, как правило, прикованы к постели.
ISH клинически похож на JHF, но отличается более тяжелым течением, неблагоприятным прогнозом и проявляется в течение первых нескольких недель или месяцев жизни ребенка. Распространенность данной патологии составляет менее 1 случая на 1 000 000 населения планеты. До 1994 года зарегистрировано 11 больных с инфантильным системным гиалинозом, и все они умерли в раннем детстве, в основном из-за тяжелой диареи, легочных инфекций и сепсиса. С тех пор было зарегистрировано более 150 случаев данной патологии. Заболевание регистрировалось в семьях различных этнических групп на нескольких континентах. В одном из отчетов описано несколько случаев среди арабского населения, что может быть связано с близкородственными браками [2, 11, 12, 20].
Уже на первом году жизни развиваются рецидивирующие гнойные инфекции, диарея и выраженный остеопороз. Интенсивные артралгии, контрактуры с ограничением объема движений в суставах приводят к неподвижности, развитию застойных явлений в легких и дыхательной недостаточности. В связи с гиалиновой инфильтрацией кишечной стенки и развивающейся энтеропатией возникает потеря белка и, как следствие, проблемы с кормлением, белково-энергетическая недостаточность, кахексия [4, 8, 15]. Смерть наступает вследствие вторичного сепсиса с полиорганной недостаточностью, обычно в возрасте до двух лет.
ISH наследуется по аутосомно-рецессивному типу, а это значит, что обе копии гена в каждой ячейке имеют мутации. Родители ребенка, каждый из которых несет по одной копии мутантного гена, как правило, не имеют симптомов заболевания. Данное заболевание связано с геном ANTXR2 (также известным как ген CMG2), расположенным на длинном плече хромосомы 4 (4q21) и кодирующим протеин-2 [9, 7, 17, 22], который участвует в образовании капилляров. Считается, что именно этот белок сохраняет нормальное строение базальных мембран, которые представляют собой структуры, поддерживающие клетки в самых разнообразных тканях. В настоящее время известно, что ген ANTXR2 является единственным, мутации в котором вызывают возникновение инфантильного системного гиалиноза. Признаки и симптомы инфантильного системного гиалиноза определяются накоплением гиалина в различных тканях организма. Природа этого вещества окончательно не установлена, предполагают, что его химический состав представлен белком и молекулами глюкозы.
Диагностика ISH основывается на совокупности клинических данных, результатов гистологического исследования пораженных тканей и генетического тестирования.
Клинические данные включают в себя 11 симптомов (представлены в порядке их диагностической специфичности).
1. Болезненные контрактуры суставов с прогрессирующим ограничением объема движения. Отмечается недостаточная активность плода во время беременности. Развиваются врожденные быстро прогрессирующие контрактуры верхних и нижних конечностей. У большинства детей уже в периоде новорожденности отмечается ограничение пассивных и/или активных движений в конечностях. Положение ребенка становится вынужденным: руки согнуты в локтевых и лучезапястных суставах и приведены к туловищу (рис. 1). Нижние конечности согнуты в коленных и голеностопных суставах. Любые попытки разгибания конечностей вызывают сильную болевую реакцию.
2. Одним из проявлений ISH является кожный синдром, характеризующийся появлением пятнисто-папулезных высыпаний, от белесоватого до розовато-жемчужного цвета, различной величины и степени интенсивности. Сыпь имеет прогрессирующий характер, что нередко приводит к появлению осложнений, таких как инфицирование и развитие вторичного сепсиса. Излюбленная локализация высыпаний — шея, волосистая часть головы, уши, руки. Кроме того, высыпания могут появиться в области половых органов и анального отверстия. При пальпации кожа твердая, плотная, складчатая, с потерей эластичности.
3. Гиперпигментация кожи: характерные багровые пятна развиваются на медиальной и латеральной поверхности лодыжек, над пястно-фаланговыми и межфаланговыми суставами кистей, в области позвоночника (рис. 2). Степень гиперпигментации изменяется в зависимости от исходной пигментации кожи.
4. Гипертрофия десен.
5. Задержка физического развития. Умственное развитие, как правило, нормальное, однако описаны единичные случаи потери когнитивных функций [12].
6. Особенности фенотипа: грубые черты лица, брахицефалия, широкое лицо, плоский затылок, голубые склеры, гипертрофия десен, хрящевая ткань на деснах, высокое небо, короткая шея, широкая и короткая грудная клетка, нанизм.
7. Чрезмерное потоотделение.
8. Гепатомегалия (может не выявляться).
9. Высокая частота переломов.
10. Высокая восприимчивость к инфекциям.
11. Развитие тяжелой диареи.
Параклинические методы диагностики
1. Иммуногистохимическое исследование биоптата кожи и слизистых: накопление гиалинового материала в дерме. Гиалин выявляется в виде аморфного эозинофильного вещества, содержащего гликопротеины и коллаген; нормальное соотношение коллагена типа I и типа III [10, 14, 18].
2. Электронное микроскопическое исследование кожи: обилие гомогенного, аморфного, эозинофильного внеклеточного матрикса, в котором встроены веретенообразные клетки, имеющие гипертрофированный аппарат Гольджи и расширенную, шероховатую эндоплазматическую сеть; высокая плотность цитоплазмы (фибробластоподобная), наличие гликопротеинов, гиалуроновой кислоты, гликозаминогликанов, хондроитинсульфатов А и С, дерматансульфата и небольшое количество тонких коллагеновых волокон [10, 14, 18, 21].
3. При выраженных желудочно-кишечных симптомах целесообразно выполнить фиброэзофагогастродуоденоскопию, ректороманоскопию с гистологическим исследованием биоптата пораженного участка слизистой оболочки кишки. В биоптатированном материале выявляют атрофию ворсинок, отек, лимфангиэктазию, гиалиноз [12].
4. Рентгенографически выявляются остеопения, периостальная реакция, остеолитические поражения и переломы костей[12, 15, 25].
5. Офтальмологический осмотр не выявляет каких-либо характерных изменений и может быть использован в дифференциальной диагностике наследственного системного гиалиноза и некоторых лизосомальных болезней накопления [12, 25].
6. При проведении биопсии мышц могут быть выявлены характерные миопатические изменения [12, 25].
7. В гемограмме определяется нормальная или незначительно повышенная скорость оседания эритроцитов (СОЭ), анемия и/или тромбоцитоз, лимфопения [12, 25].
8. Дефицит гуморального и клеточного звеньев иммунитета (гипогаммаглобулинемия, снижение уровня IgG и IgA, IgM в пределах нормы) [6, 24].
Инфантильный системный гиалиноз следует дифференцировать с липогранулематозом Фарбера, липоидным протеинозом Урбаха — Вите (proteinosis lipoidica Urbach-Wiethe), синдромом Винчестера (Winchester), мукополисахаридозом II типа (синдром Хантера).
В связи с отсутствием специфического лечения инфантильного системного гиалиноза терапия направлена на улучшение качества жизни и включает физиотерапевтические и симптоматические средства, профилактику вторичных осложнений [2, 4, 15, 21]. Около 80 % детей умирают в течение первых двух лет жизни, в основном из-за рецидивирующих инфекций дыхательных путей и тяжелой диареи. У пациентов, которые доживают до более старшего возраста, мобильность ограничена за счет жестких контрактур.
В качестве иллюстрации инфантильного системного гиалиноза предлагаем вашему вниманию клинический случай (собственное наблюдение).
Мальчик С., 1 года 3 месяцев, госпитализирован в связи с развитием контрактур локтевых, коленных суставов, суставов кистей, стоп, резкой болезненностью и ограничением движений в них, явлениями дерматита, задержкой психомоторного и речевого развития.
Из анамнеза болезни известно, что у ребенка с рождения отмечались гипотония верхних конечностей и ограничение разгибания нижних конечностей в коленных суставах, по поводу чего ребенок неоднократно консультирован ортопедом, неврологом, проходил курсы массажа. В возрасте 3 месяцев госпитализирован по месту жительства с диагнозом: последствия внутриутробной и перинатальной патологии; спастико-тонический синдром, двигательные расстройства; дисплазия тазобедренных суставов. На фоне восстановительного лечения (ЛФК, массаж, иммобилизация конечностей шиной, электростимуляция на верхние и нижние конечности) состояние частично улучшилось: появились активные движения в верхних и нижних конечностях, но не в полном объеме. В возрасте 5 месяцев у ребенка появилась припухлость в области локтевых, лучезапястных суставов, суставов кистей, стоп, коленных, голеностопных суставов, выраженный болевой синдром, багрово-синюшное окрашивание кожи над суставами, ограничение движения в них. К 8-му месяцу присоединился кожный синдром в виде красновато-синюшного утолщения и уплотнения кожи в области шеи, спины, мелких папулезных высыпаний на лице, гиперплазии десен; изменения слизистых в виде гипертрофии альвеолярных отростков верхней челюсти. В возрасте 9 месяцев ребенок консультирован ортопедом в Институте патологии позвоночника и суставов имени проф. Н.И. Ситенко, высказано предположение о болезни накопления (возможно, мукополисахаридоз). Констатированы дисплазия тазобедренных суставов, контрактуры суставов верхних и нижних конечностей, остеопороз; консолидированные переломы проксимальных отделов обеих бедренных костей, костей левого предплечья. Для верификации диагноза ребенок направлен в медико-генетический центр г. Харькова, где на основании клинических данных установлен диагноз: липогранулематоз Фарбера. Нарушение обмена серосодержащих аминокислот. Гипергомоцистеинемия. Вторичная митохондриальная дисфункция. Тип наследования аутосомно-рецессивный.
Из анамнеза жизни: ребенок от первой нормально протекавшей беременности. Роды первые, срочные, путем кесарева сечения, в ягодичном предлежании. Масса тела при рождении — 3050 г, длина — 50 см. Из роддома ребенок выписан на 5-е сутки в удовлетворительном состоянии. Наследственность не отягощена.
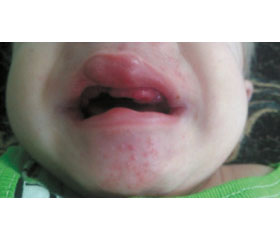
При объективном обследовании состояние ребенка тяжелое за счет основного заболевания. В фенотипе обращают на себя внимание грубые черты лица, широкое лицо, гиперплазия десен, высокое небо, короткая шея, широкая короткая грудная клетка, контрактуры локтевых, коленных суставов, суставов кистей, пигментация кожи над суставами, гиперпигментация мошонки, на руках «четки», гигромы кистей, хондромы стоп, локтей, папиллома правого уха, гиперемия участков кожи волосистой части головы, спины, крестца, мелкая папулезная сыпь на лице (рис. 3–5).
Тургор кожи удовлетворительный. При аускультации легких выслушивалось везикулярное дыхание. Границы сердца в пределах возрастной нормы. Тоны сердца ясные, чистые, звучные. Живот обычных размеров, мягкий, безболезненный при пальпации. Печень, селезенка не увеличены. Стул оформленный. Мочеиспускание свободное. Видимая задержка в физическом, речевом развитии. Интеллект сохранен.
В общем анализе крови: эритроциты — 5,46 • 1012/л, гемоглобин — 84 г/л, лейкоциты — 14,12 • 109/л, палочкоядерные нейтрофилы — 1 %, сегментоядерные нейтрофилы — 36 %, лимфоциты — 51 %, моноциты — 10 %, тромбоциты — 769,0 • 106/л, СОЭ — 15 мм/ч. В биохимическом анализе крови выявлено снижение уровня сывороточного железа до 3,3 мкмоль/л, повышение уровня С-реактивного белка (СРБ) до 6,0 г/л, альфа-фетопротеина — до 4,19 МE/мл (норма < 2,64).
По данным рентгенографии тазобедренных суставов, бедер, голеней, кистей выявлен выраженный равномерный остеопороз. В области суставов визуализируются дополнительные мягкотканные образования. Поднадкостничный перелом метафиза правого бедра, проксимального метафиза левой большеберцовой кости с образованием выраженной реакции консолидации (аналогичные изменениям при osteogenesis imperfecta). Остеогенез соответствует возрасту.
Рентгенография позвоночника, черепа, органов грудной клетки не выявила патологических изменений.
По данным компьютерной томографии позвоночника, верхних и нижних конечностей отмечается незаращение дуги L5, а также дуг всех крестцовых позвонков. В области больших суставов — дополнительные мягкотканные образования. Определяются переломы проксимальных отделов обеих бедренных костей с признаками выраженного периостального и эндостального костеобразования. По данным магнитно-резонансной томографии голеностопных суставов соотношение в суставах не нарушено. Суставные хрящи прослеживаются на всем протяжении, без видимых структурных изменений. Значительное количество жидкости в полостях голеностопных и таранно-пяточных суставов с обеих сторон. На уровне голеностопных суставов определяются отек и утолщение длинных разгибателей и сгибателей пальцев, коротких малоберцовых мышц с обеих сторон.
УЗИ органов брюшной полости, компьютерная томография органов грудной клетки и брюшной полости не выявили патологических изменений.
По данным ЭхоКГ отмечалось незначительное увеличение полости левого желудочка. Сократимость миокарда хорошая. Стенки левого желудочка незначительно утолщены. Визуализируется перимембранозный дефект межжелудочковой перегородки диаметром 4 мм, частично прикрыт структурами трикуспидального клапана, без признаков легочной гипертензии. Минимально открытое овальное окно.
Электрокардиограмма: метаболические изменения в миокарде.
С учетом данных клинико-лабораторного, инструментального обследования проводилась дифференциальная диагностика между болезнями накопления, липогранулематозом Фарбера, генетическими заболеваниями, туберкулезным поражением костно-суставной системы.
Для верификации диагноза проведена биопсия пораженной кожи в области спины, латеральной лодыжки левой нижней конечности. Патогистологическое заключение: фрагмент кожи и мягких тканей с коллагеновым фиброзом, очагами гиалиноза; участки изъязвления с воспалительной реакцией в подлежащих тканях и наличием капиллярных синусоидов.
Таким образом, подтвержден диагноз: инфантильный системный гиалиноз, тип наследования аутосомно-рецессивный. Повторный генетический риск в данном браке 25 %.
За время пребывания в стационаре состояние ребенка оставалось тяжелым по основному заболеванию, развился интестинальный синдром, лабораторно сохранялись признаки анемии смешанного генеза, невысокий лейкоцитоз с лимфомоноцитарным преобладанием, высокие показатели СРБ. Пациент получал симптоматическую, дезинтоксикационную и регидратационную терапию.
Описанный клинический случай инфантильного семейного гиалиноза — первый на территории Украины. Данная патология относится к группе орфанных болезней, развивается преимущественно в раннем детском возрасте и быстро прогрессирует, приводя к тяжелой инвалидизации, часто — с фатальными инфекционными осложнениями. Все вышеизложенное требует от врача-педиатра знания генетической синдромной патологии, необходимости мультидисциплинарного подхода в диагностике данного заболевания.
Список литературы
1. Adams S. Mutations in the gene encoding capillary morphogenesis protein 2 cause juvenile hyaline fibromatosis and infantile systemic hyalinosis / Adams S., Hanks S., Douglas J., Arbour L., Atherton D.J., Balci S., Bode H., Campbell M.E., Feingold M., Keser G., Kleijer W., Mancini G., McGrath J.A., Muntoni F., Nanda A., Teare M.D., Warman M., Pope F.M., Superti-Furga A., Futreal P.A., Rahman N. // Am. J. Hum. Genet. — 2003. — Vol. 73, № 4. — P. 791-800. — Doi:http://dx.doi.org/10.1086/378418.
2. Al-Mayouf S.M. Infantile systemic hyalinosis: a fatal disorder commonly diagnosed among Arabs / Al-Mayouf S.M., Al-Mehai–dib A., Bahabri S., Shabib S., Sakati N., Teebi A.S. // Clin. Exp. Rheumatol. — 2005. — Vol. 23, № 5. — P. 717-720. — PMID: 16173255.
3. Al-Mubarak L. Infantile systemic hyalinosis presenting as intractable infantile diarrhea / Al-Mubarak L., Al-Makadma A., Al-Khenaizan S. // Eur. J. Pediatr. — 2009. — Vol. 168, № 3. — P. 363-365. — Doi:10.1007/s00431-008-0760-8.
4. Büyükgebiz B. A rare cause of protein-losing enteropathy and growth retardation in infancy: infantile systemic hyalinosis / B. Büyükgebiz, Y. Ӧztürk, N. Arslan, E. Оzer // Turk. J. Pediatr. — 2003 Jul-Sep. — Vol. 45, № 3. — P. 258-260.
5. Criado G.R. Infantile systemic hyalinosis: a clinicopathological study / Criado G.R., Gonzаlez-Meneses A., Caсadas M., Rafel E., Yanes F., De Terreros I.G. // Am. J. Med. Genet. A. — 2004. — Vol. 129A, № 3. — P. 282-285. — PMID: 15326628.
6. De G.C. Administration of intravenous immunoglobulin in two children with hypogammaglobulinaemia due to protein losing entero–pathy / De G.C., Maggiore G., Scotta M.S., Ugazio A.G. // Clin. Exp. Immunol. — 1985. — 60. — P. 447-448. — PMID: 4006307.
7. Denadai R. Identification of 2 novel ANTXR2 mutations in patients with hyaline fibromatosis syndrome and proposal of a modified grading system / Denadai R., Raposo-Amaral C.E., Bertola D., Kim C., Alonso N., Hart T., Han S., Stelini R.F., Buzzo C.L., Raposo-Amaral C.A., Hart P.S. // Am. J. Med. Genet. A. — 2012 Apr. — 158 A(4). — P. 732-742. — Doi: 10.1002/ajmg.a.35228. Epub 2012 Mar 1.
8. Dhingra M. Juvenile hyaline fibromatosis and infantile systemic hyalinosis: Divergent expressions of the same genetic defect? / –Dhingra M., Amladi S., Savant S., Nayak C. // Indian. J. Dermatol. Venereol. Leprol. — 2008. — 74. — P. 371-374. — http://imsear.hellis.org/handle/123456789/52180
9. El-Kamah G.Y. Spectrum of mutations in the ANTXR2 (CMG2) gene in infantile systemic hyalinosis and juvenile hyaline fibromatosis / El-Kamah G.Y., Fong K., El-Ruby M., Afifi H.H., Clements S.E., Lai-Cheong J.E., Amr K., El-Darouti M., McGrath J.A. // Br. J. Dermatol. — 2010. — Vol. 163, № 1. — P. 213-215. — Doi: 10.1111/Дж.1365-2133.2010.09769.х
10. Ihikawa H. Systemic hyalinosis (juvenile hyaline fibromatosis). Ultrastructure of the hyaline with particular reference to the cross-banded structure / Ishikawa H., Maeda H., Takamatsu H., Saito Y. // Arch. Dermatol. Res. — 1979 Jun 25. — Vol. 265, № 2. — P. 195-206. — PMID: 88923.
11. Jaouad I.C. Consanguineous marriages in Morocco and the consequence for the incidence of autosomal recessive disorders / Jaouad I.C., Elalaoui S.C., Sbiti A., Elkerch F., Belmahi L., Sefiani A. // J. Biosoc. Sci. — 2009 Sep. — 41(5). — P. 575-81. — Doi: 10.1017/S0021932009003393. Epub 2009 May 12.
12. Joseph T.C. Shieh. Hyalinosis, Inherited Systemic Synonym: Hyaline Fibromatosis Syndrome / Joseph T.C. Shieh, E. Hoyme, L.T. Arbour // Initial Posting. — February 27, 2008. — Last Update. — April 11, 2013. — PMID: 20301698.
13. Kawasaki G. Juvenile hyaline fibromatosis complicated with oral squamous cell carcinoma: a case report / Kawasaki G., Yanamoto S., Mizuno A., Fujita S. // Oral. Surg. Oral. Med. Oral. Pathol. Oral. Radiol. Endod. — 2001 Feb. — Vol. 91, № 2. — P. 200-204. — PMID: 11174598.
14. Mayer-da-Silva. Juvenile hyaline fibromatosis. A histologic and histochemical study / Mayer-da-Silva, Poiares-Baptista A., Guerra Rodrigo F., Teresa-Lopes M. // Arch. Pathol. Med. — 1988 Sep. — № 112(9). — P. 928-931.
15. Nischal K.C. Juvenile hyaline fibromatosis / Nischal K.C., Sachdev D., Kharkar V., Mahajan S. // J. Postgrad. Med. — 2004. — Vol. 50, № 2. — P. 125-126. — PMID: 15235211.
16. Nofal A. Juvenile hyaline fibromatosis and infantile systemic hyalinosis: a unifying term and a proposed grading system / Nofal A., Sanad M., Assaf M., Nofal E., Nassar A., Almokadem S., Attwa E., Elmosalamy K. // J. Am. Acad. Dermatol. — 2009 Oct. — 61(4). — P. 695-700. — Doi: 10.1016/j.jaad.2009.01.039. Epub 2009 Apr 2.
17. Rahman N. The gene for juvenile hyaline fibromatosis maps to chromosome 4q21 / Rahman N., Dunstan M., Teare M.D., Hanks S., Edkins S.J., Hughes J., Bignell G.R., Mancini G., Kleijer W., Campbell M., Keser G., Black C., Williams N., Arbour L., Warman M., Superti-Furga A., Futreal P.A., Pope F.M. // Am. J. Hum. Genet. — 2002. — Vol. 71, № 4. — P. 975-980. — PMID: 12214284.
18. Remberger K. Fibromatosis hyalinica multiplex (juvenile hyalin fibromatosis). Light microscopic, electron microscopic, immunohistochemical, and biochemical findings / Remberger K., Krieg T., Kunze D., Weinmann H.M., Hubner G. // Cancer. — 1985 Aug 1. — 56(3). — P. 614-624.
19. Shieh J.T.C. Systemic hyalinosis: a distinctive early childhood-onset disorder characterized by mutations in the anthrax toxin receptor 2 gene (ANTXR2) / Shieh J.T.C., Swidler P., Martignetti J.A., Ramirez M.C., Balboni I., Kaplan J., Kennedy J., Abdul-Rahman O., Enns G.M., Sandborg C., Slavotinek A., Hoyme H.E. // Pediatrics. — 2006. — Vol. 118, № 5. — P. e1485-1492. — PMID: 17043134.
20. Siham Al Sinani. Infantile Systemic Hyalinosis: A Case Report with a Novel Mutation / Siham Al Sinani, Fathyia Al Murshedy, Reem Abdwani // Oman. Med. J. — 2013 Jan. — 28(1). — P. 53-55. — Doi: 10.5001/omj.2013.12.
21. Stucki U. Infantile systemic hyalinosis in siblings: clinical report, biochemical and ultrastructural findings, and review of the literature / Stucki U., Spycher M.A., Eich G., Rossi A., Sacher P., Steinmann B., Superti-Furga A. // Am. J. Med. Genet. — 2001. — № 100. — P. 122-129. — Doi: 10.1002/1096-8628(20010422)100:2<122::AID-AJMG1236>3.0.CO;2-0
22. Urbina F. Infantile systemic hyalinosis or juvenile hyaline fibromatosis? / Urbina F., Sazunic I., Murray G. // Pediatr. Dermatol. — 2004. — Vol. 21, № 2. — P. 154-159. — PMID: 15078358.
23. Whitfield A., Robinson A.H. A further report on the remarkable series of cases of molluscum fibrosum in children communicated to the Society by Dr. John Murray in 1873 // Med. Chir. Trans. — 1903. — 86. — P. 293-302. — PMCID: PMC2037211.
24. Yana Klebanova, Christina Schwindt. Infantile Systemic Hyalinosis: A Case Report of Compromised Cellular and Humoral Branches of the Immune System Leading to Infections // Pediatr. Asthma Allergy Immunol. — 2009 Sep. — 22(3). — P. 127-130. — Doi: 10.1089/pai.2009.0011
25. Zolkipli Z. Skeletal muscle involvement in infantile systemic hyalinosis / Zolkipli Z., Longman C., Brown S., Rahman N., Holder S.E., Muntoni F. // Eur. J. Paediatr. Neurol. — 2003. — Vol. 7, № 6. — P. 401-406. — PMID: 14623219.

/113.jpg)
/114.jpg)