Статья опубликована на с. 118-123
Слуга Джо, один із персонажів роману англійського письменника Чарлза Діккенса «Посмертні записки Піквікського клубу», — «жирний, червонопикий чоловік, занурений у дрімоту», якого в романі називають «міхуром» (через ожиріння), «юним споживачем опіуму» (за сонливість) і «удавом» (через ненажерливість). Можливо, це перший опис синдрому Прадера — Віллі?
Історичні відомості
Мабуть, уперше синдром Прадера — Віллі (СПВ) було описано в дівчини-підлітка Дж. Ленгтоном Дауном у 1887 році, але цей випадок не згадувався в медичній літературі протягом 70 років. Дівчина мала розумову відсталість, ожиріння, низький зріст, гіпогонадизм. Сукупність цих симптомів об’єднали під назвою «полісаркія». У 1956 році швейцарські лікарі Андреа Прадер, Алексис Лабхарт і Генріх Віллі висловили припущення про автосомно-рецесивний тип успадкування захворювання. Потім з’явилися повідомлення про можливість автосомно-домінантної передачі хвороби [5]. Підтвердженням даних гіпотез служили сімейні випадки дев’яти хворих (п’ять хлопчиків і чотири дівчини віком від 5 до 23 років) зі схожими клінічними проявами, притаманними даній патології [12]. З 1970 року цей розлад отримав назву синдрому Прадера — Віллі [2].
Поширеність. За даними Глобального реєстру, синдром Прадера — Віллі — це комплексне мультисистемне генетичне захворювання, що трапляється приблизно в одного з кожних 15 000 новонароджених, з однаковою частотою серед чоловіків та жінок незалежно від раси чи народності.
Генетичні аспекти. Народження хворої дитини обумовлено делецією батьківської копії імпринтованої ділянки проксимального відділу довгого плеча 15-ї хромосоми (15q11-q13) [1]. Так званий PWS/AS-регіон може бути втрачений у результаті дії одного з кількох генетичних механізмів, у більшості випадків через мутації. Як правило, людина отримує по одній копії 15-ї хромосоми від матері та батька. Через дію імпринтингу активність копій вищезазначених генів, що були успадковані від матері, дуже низька або взагалі відсутня, тобто виражені тільки батьківські копії генів. СПВ виникає в результаті втрати активності батьківської копії цього регіону.
Генетичний дефект може виникнути в одному з трьох напрямків. Найчастіше спостерігається видалення батьківської хромосоми 15q11-q13. Ця мала делеція виникає в 70 % випадків та зазвичай не виявляється за допомогою звичайного генетичного аналізу, такого як каріотипування при амніоцентезі [5, 9].
У 20–30 % хворих на синдромом Прадера — Віллі, які мають за даними цитогенетичного дослідження нормальний каріотип, за допомогою молекулярно-біологічних методів виявляється уніпарентна материнська дисомія 15-ї хромосоми. Батьківська 15-та хромосома у таких хворих відсутня. Ця своєрідна «гомозиготизація» відбувається в результаті нерівного кросинговеру або соматичної рекомбінації за механізмом конверсії генів. Дуже мала частка (1–3 %) випадків обумовлена генетичною мутацією, що спричинена інактивацією генетичного матеріалу батьківської 15-ї хромосоми [5].
Цікаво, що якщо делеції відбуваються в цьому ж регіоні на материнській хромосомі, то це призводить до виникнення синдрому Ангельмана. Приблизно 60 % пацієнтів із синдромом Ангельмана мають таку саму делецію (15ql1-q13), що виникає в материнській успадкованій хромосомі. Патернальна уніпарентальна дисомія трапляється в 3–5 % випадків. У приблизно 10 % наявна імпринтована мутація, а в інших випадках категорія хворих, що не описана у СПВ, має презюмовану одногенну мутацію. Для синдрому Ангельмана притаманні такі клінічні прояви, як рання (у віці 6–12 місяців) затримка психомоторного розвитку, мікроцефалія, порушення мовлення (у 100 % випадків), атаксія, нестримний сміх, часті епілептиформні напади. Синдром Прадера — Віллі та синдром Ангельмана — це перші описані випадки порушення процесу імпринтингу людини [8].
Останнім часом відзначаються деякі клінічні відмінності між пацієнтами з мікроделецією батьківського походження та материнською ізодисомією. Це пов’язано з відсутністю типового фенотипу хворих з уніпарентною материнською дисомією, що значно затримує своєчасну постановку діагнозу. Особи афроамериканського походження з діагнозом СПВ також фенотипово відрізняються: зазвичай хворі нормального зросту, розміри кистей та стоп відповідають розмірам тіла [6].
Клінічні особливості й ознаки
Хоча цей синдром має багато специфічних ознак, таких як інфантильна м’язова гіпотонія, затримка фізичного розвитку та розумова відсталість, гіпогонадотропний гіпогонадизм, поведінкові розлади, характерний вираз обличчя, низький зріст та акромікрія, остаточний діагноз встановлюється з появою гіперфагії та прогресивним розвитком ожиріння. Доктор Батлер (Butler) виявив, що на момент постановки діагнозу СПВ середній вік пацієнтів становив понад 6 років [8].
Існує думка щодо появи патогномонічних проявів синдрому внаслідок функціональної недостатності гіпоталамусу (низький зріст, центральне ожиріння, гіпогонадизм та остеопороз) [2], але клінічна картина відрізняється залежно від віку пацієнта. Тому впродовж усього періоду захворювання можна виділити 2 фази. Перша властива дітям неонатального віку та періоду раннього дитинства. Вона характеризується різним ступенем м’язової гіпотонії, недостатньою масою тіла, слабким криком, зниженням рефлексів (Моро, смоктального, ковтального), що ускладнює годування дитини та іноді вимагає розміщення шлункового зонду або накладання гастростоми, густою слиною, затримкою статомоторного розвитку, температурною нестабільністю, гіпоплазією статевих органів (крипторхізм, гіпоплазія калитки, гіпоплазія клітора). Гіпотонія центрального генезу поступово зменшується до 8–11-го місяця. Дитина пізно починає тримати голову, сидіти (близько 11–12 місяців від народження), повзати (у 15–16 місяців), ходити (у 24–27 місяців), говорити (у 38–39 місяців) [6, 16, 18].
Недостатня маса тіла у віці 6 місяців — 6 років змінюється прогресивним розвитком ожиріння переважно на тулубі і в проксимальних відділах кінцівок унаслідок гіперфагії, що є патогномонічним проявом другої фази захворювання. Зазвичай цей етап починається приблизно у два роки та характеризується затримкою росту; кінцевий зріст за відсутності лікування для дорослих становить 155 см для чоловіків та 147 см для жінок. До основних клінічних проявів другого етапу належать помірна розумова відсталість, поведінкові розлади — різка зміна настрою, істерики, упертість, нав’язливо-спонукальна (обсесивно-компульсивна) поведінка; харчова поведінка характеризується постійним пошуком їжі, вживанням будь-яких продуктів, навіть брудних [4]. Інші ознаки другого етапу включають аномалії окорухового апарату (косоокість, ектропіон, глаукома); патологію ротової порожнини (мікродонтія, гіпоплазія емалі, карієс зубів, неправильний прикус, густа слина); розлади артикуляції, немотивовану сонливість, адинамію, знижену больову чутливість, гіпотермію, сколіоз, апное під час сну, гіпопігментацію волосся, шкіри та райдужки [6, 16].
Курф і Фрим (Curfs та Frym) у 1992 році вивчали різноманітні ступені розумових відхилень та труднощів при навчанні серед осіб, хворих на СПВ. Результати їхнього дослідження були такими: 5 % мають середній рівень коефіцієнта інтелекту; 27 % — на межі середнього; 39 % мають незначну розумову відсталість; 27 %— помірну розумову відсталість; 1 % — тяжку розумову відсталість; < 1 % — глибоку розумову відсталість [4, 13].
Як правило, діти з синдромом Прадера — Віллі мають хорошу довготривалу зорову пам’ять, вони можуть навчитися читати, мають багатий пасивний словниковий запас, проте їхні мовленнєві здібності суттєво поступаються їхньому розумінню. Слухова та зорова короткострокова пам’ять, звукова концентрація уваги знаходяться на досить низькому рівні, так само як здібності до точних дисциплін і письма [4, 13].
Характерні риси обличчя: високий та вузький лоб, мигдалеподібні очі з тонкими, опущеними вниз повіками, великий та широкий ніс із вузькою спинкою носа, відкритий рот із тонкою верхньою губою, гіпопігментація шкіри, волосся, рогівки, страбізм. Маленькі кисті та ступні зазвичай помітно у віці 10 років, середня довжина ступні дорослої жінки — 20,3 см, чоловіка — 22,3 см [16].
Діагностика
У 1993 році Холм (Holm), Кессіді (Cassidy), Батлер (Butler) розробили схему діагностичних критеріїв (головні, мінімальні та додаткові) СПВ. Основний критерій оцінюється в один бал, мінімальний оцінюється у півбала. Додатковий критерій не оцінюється, але може бути підтвердженням діагнозу генетичної хвороби. У дітей віком до 3 років діагноз СПВ клінічно діагностується за наявності 5 ознак (3 — з числа головних критеріїв та 2 — з мінімальних), у дітей понад 3 роки — за наявності 8 ознак (4 головних + 4 мінімальних) [18].
Діагностичні критерії синдрому Прадера — Віллі
Головні критерії
— ЦНС: м’язова гіпотонія в немовляти.
— Шлунково-кишковий тракт: проблеми годування і/або затримка фізичного розвитку в немовляти.
— Харчування: швидка прибавка маси тіла у дитини віком 6 місяців — 6 років.
— Черепно-лицеві дизморфії: вузький лоб, мигдалеподібний розріз очей, вузька спинка носу, опущені кути рота.
— Ендокринні: гіпогонадизм.
— Розвиток: затримка розвитку та/або розумова відсталість.
Мінімальні критерії
— Неврологічні: зменшення рухової активності плода/інфантильна летаргія.
— Дихальні: порушення сну і/або апное під час сну.
— Ендокринні: низький очікуваний ріст (нижче середнього дорослого росту).
— Дерматологічні: гіпопігментація шкіри та волосся.
— Ортопедичні: маленькі кисті і ступні.
— Ортопедичні: вузькі руки з ульнарною девіацією.
— Офтальмологічні: страбізм/міопія.
— Стоматологічні: густа клейка слина, карієс.
— ЛОР: дефекти мовленнєвої артикуляції, дизлалія.
— Психіатричні: пацієнти можуть роздирати, рвати власну шкіру (у деяких випадках у хворих із СПВ розвивається анемія внаслідок вторинної кишкової кровотечі після травмування шкіри в ділянці анусу).
Додаткові критерії
— Неврологічні: високий больовий поріг і нормальні при гіпотонії нейром’язові показники.
— Гастроентерологічні: зменшення блювання.
— Ендокринологічні: неефективна терморегуляція, раннє adrenarche і/або остеопороз.
— Ортопедичні: сколіоз/кіфоз.
— Загальний розвиток: вміло складають картинки-пазли.
Для уточнення діагнозу синдрому Прадера — Віллі необхідно провести генетичне тестування [9, 14, 18]. Це тестування включає в себе хромосомний аналіз (молекулярно-цитогенетичне дослідження, каріотипування) для виявлення мікроделеції 15-ї хромосоми в локусі 15q11.2-q13 та оцінку процесів метилювання в локусі (SNRPN and PW71) за допомогою блот-гібридизації за Саузерном або полімеразної ланцюгової реакції (ПЛР) із використанням ДНК-праймерів для виявлення метилювання цитозину. Здорові особи мають обидві (метильовану та неметильовану) алелі, у той час як особи з СПВ мають лише материнську метильовану алель. Цитогенетичний аналіз високої точності може виявити делецію 15q11-q13; хоча зберігається великий відсоток отримання хибно-негативного або хибно-позитивного результату. Флюоресценція в гібридизації з використанням зондів у межах критичної ділянки СПВ (SNRPN чи DI 5SI 1), є делеції звичайного розміру, що спричиняє СПВ. Уніпарентна дисомія може бути виявлена за допомогою ПЦР із використанням мікросателітних маркерів із ділянки СПВ. Аналіз детекції уніпарентної дисомії потребує зразків від обох батьків та дитини із СПВ.
Пренатальна діагностика
Пренатальна діагностика СПВ можлива. Метод флюоресцентної гібридизації in situ (FISH) може бути визначальним діагностичним тестом для підтвердження пренатального діагнозу делеції в ділянці 15q під час проведення біопсії ворсин хоріону або амніоцентезу. Ризик народження дитини з СПВ у сім’ї, де вже є один хворий нащадок, залежить від механізму виникнення генетичної хвороби. Ймовірність народження хворої дитини становить менше 1 %, якщо цей випадок обумовлений делецією гена або уніпарентною дисомією. У пацієнтів із мутацією в центрі імпринтингу тестуються біологічні батьки на наявність асимптомних мутацій у центрі імпринтингу; вказані мутації зумовлюють високий повторний ризик синдрому (50 %). У випадку появи хромосомних транслокацій ризик виникнення розладу в наступної дитини становить 25 % [11, 14].
Ускладнення
Ускладнення, що виникають унаслідок ожиріння, — апное під час сну, легенево-серцева недостатність, гіпертензія, тромбофлебіт, цукровий діабет 2-го типу, вивих голівки стегна та хронічний набряк ніг, — є головними причинами високої захворюваності та смертності осіб із СПВ. Дослідження Лемба (Lamb) свідчать про передчасний розвиток атеросклерозу коронарних судин, патологічного ожиріння та інсулінзалежного цукрового діабету в пацієнтів із СПВ у молодому віці. Задуха стала причиною смерті у 7,9 % пацієнтів серед 152 досліджуваних осіб із даною патологією.
Інше дослідження 8 дітей та 2 дорослих описує синдром раптової смерті як одну з причин смертності; у 3 із 8 дітей відмічають невеликі розміри надниркових залоз, що свідчить про надниркову недостатність. Подальші дослідження оскаржують частоту центральної надниркової недостатності, запропоновану цими авторами, вважаючи, що це рідкісний випадок [11].
Прогноз
СПВ вважається рідкісним генетичним розладом, але найбільш частою відомою генетичною формою дитячого ожиріння [2]. Популяційна частота патології становить 350 000–400 000 людей в усьому світі. На жаль, це захворювання не завжди діагностується вчасно, незважаючи на різноманітність діагностичних критеріїв. Метою цієї статті є надання додаткової інформації стосовно СПВ, а також підвищення рівня виявлення СПВ у роботі практикуючих педіатрів. Своєчасна постановка діагнозу СПВ справляє значний вплив на здоров’я та якість життя осіб із діагнозом СПВ. Контроль за характерним ожирінням та розладами поведінки є серйозним завданням для педіатрів, генетиків, ендокринологів, дієтологів, психіатрів та піклувальників.
Сучасні методи лікування
Згідно з наказом МОЗ України № 55 про надання медичної допомоги дітям із СПВ від 03.02.2009, терапія повинна бути спрямована в 6 основних напрямках: початкова корекція м’язової гіпотонії або проблем вигодовування, лікування ожиріння, затримки росту, гіпогонадизму або гіпопітуїтаризму, поведінкових проблем, лікування інших ускладнень та проявів СПВ (цукрового діабету, апное під час сну, синдрому Піквіка), контроль сколіозу, корекція страбізму та ін. [17].
Ожиріння. На даний момент традиційна терапія патологічних станів, що супроводжують синдром, неефективна. Однак деякі лікувальні заходи позитивно впливають на стан здоров’я пацієнтів. Для профілактики основної причини захворюваності — ожиріння — рекомендована збалансована гіпокалорійна дієта (1000 калорій із доповненням вітамінами і кальцієм) з раннього шкільного віку під ретельним контролем дієтолога. Сьогодні не доведена ефективність препаратів з анорексигенною дією на гіперфагію в дітей із СПВ.
Лікування препаратами гормона росту зменшує масу тіла, корегує остеопенію, запобігає розвитку сколіозу і в деяких хворих корегує зміни поведінки. Вимірювання натщесерце в сироватці крові рівня зростання інсуліноподібного фактора 1 і інсуліноподібного фактора зв’язуючого білка 3 є скринінговим методом для визначення базового дефіциту гормона росту. 20 червня 2000 року управління продовольства і медикаментів США схвалило застосування препарату гормона росту в дітей із генетично підтвердженим діагнозом синдрому Прадера — Вілі. В Україні його рекомендовано вводити під шкіру щодня або 6 днів на тиждень. Сумарна тижнева доза становить 0,15–0,3 мг/кг із подальшою корекцією ефективної дози. Згідно з даними американських учених, рекомендується використовувати генотропін у дозі 0,24 мг/кг/тиждень, попередньо розділений на рівні дози для 6–7 щоденних ін’єкцій, і омнітропін у дозі 0,24 мг/кг/тиждень по 6–7 щоденних ін’єкцій. Ефективність та безпека довгострокового застосування гормона росту були оцінені в численних дослідженнях, присвячених розладам, що супроводжуються низькорослістю. Показанням для початку лікування соматотропним гормоном є доведена тенденція до затримки зросту. За літературними даними, у світовій практиці лікування розпочинають у віці від двох до чотирьох років. На сьогодні в Україні державна програма із забезпечення дітей гормоном росту поширюється на дітей із гіпофізарним нанізмом та синдромом Тернера [10, 16, 17].
Фізіотерапія. У зв’язку з наявністю гіпотонії діти потребують додаткової терапії — масаж і ЛФК для набуття основних і тонких моторних навиків, підсилення спинної мускулатури для мінімізації сколіозу. Заохочення до фізичної діяльності важливе для корекції маси тіла.
Хірургічне лікування. Хворі на СПВ можуть потребувати хірургічного лікування ускладнень ожиріння, крипторхізму та сколіозу. Вони можуть потребувати ретельного хірургічного спостереження у зв’язку з абдомінальними проблемами: внаслідок високої толерантності до болю й низької здатності до блювання можуть пізно з’являтись симптоми холециститу, апендициту або гострого розширення шлунка з ризиком прогресування до некрозу [16].
Клінічне спостереження
Пацієнтам із СПВ необхідна консультація таких фахівців [17]:
— генетика — для первинної медико-генетичної діагностики з використанням клінічних та специфічних генетичних методів дослідження та консультування з питань інформатизації щодо визначення ризику народження хворих дітей у сім’ї;
— ендокринолога — для регулярного контролю показників фізичного розвитку, оцінки досліджених лабораторних та інших показників моніторингу лікування;
— дієтолога — для консультування з приводу надлишкової ваги;
— офтальмолога — для корекції косоокості;
— пульмонолога — для профілактики виникнення апное під час сну;
— психіатра, психолога або обох фахівців — із приводу корекції поведінкових проблем;
— гастроентеролога — з питань діагностики та усунення проблем із боку шлунково-кишкового тракту.
СПВ — мультисистемний розлад із великим різноманіттям клінічних проявів. Динаміка розвитку дитини дозволяє дійти висновку, що впровадження запропонованого комплексу лікувально-педагогічних заходів може сприяти ефективному лікуванню дітей із синдромом Прадера — Віллі і можливості їх соціальної інтеграції в суспільство.
Клінічний випадок
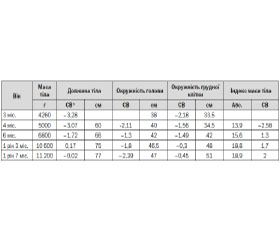
Наводимо клінічний випадок пацієнта з синдромом Прадера — Віллі з нашої практики. У червні 2013 року на консультативний прийом звернулися батьки з дитиною (хлопчик віком 4 місяці) зі скаргами на відставання в моторному та фізичному розвитку, виражену м’язову гіпотонію, проблеми з вигодовуванням та часте зригування. З анамнезу відомо, що дитина від 2-ї вагітності на фоні центрального передлежання плаценти, плановий кесарів розтин на 37–38-му тижні, народився з малою масою тіла — 2540 г. З народження спостерігалася виражена м’язова гіпотонія, відсутність смоктального рефлексу, у зв’язку з чим він знаходився на зондовому вигодовуванні впродовж 1-го місяця. Вигодовування штучне. У віці 3 місяців проходив обстеження та лікування на базі неврологічного відділення міської лікарні, де було висловлено підозру на спінальну м’язову атрофію 1-го типу, проведено відповідне генетичне тестування, що не підтвердило попередній діагноз. Проведено генетичне консультування та каріотипування (46XY). Консультований ортопедом: дисплазія кульшових суглобів.
При огляді звертали на себе увагу виражена м’язова гіпотонія, відставання в моторному (не утримував голову, не перевертався) та фізичному розвитку (табл. 1), множинні малі аномалії розвитку (близько посаджені очі, сплощене перенісся, неправильна форма голови, сандалевидна щілина, двобічний крипторхізм). Шкіра бліда, волосся світле. Реакція на огляд пасивна, але погляд фіксує, стежить за предметами.
/122.jpg)
Спираючись на дані анамнезу та результати обстеження, ми виділили такі основні патологічні симптоми: м’язова гіпотонія, відсутність смоктального рефлексу та проблеми з вигодовуванням, мала маса тіла при народженні та повільне збільшення ваги впродовж перших місяців життя, лицеві дизморфії, двобічний крипторхізм, що відповідають головним міжнародним критеріям, характерним для СПВ. Хлопчику рекомендовано консультацію генетика, УЗД органів черевної порожнини та нирок, дослідження рівня гормонів щитоподібної залози, глюкози, проведення печінкових проб, ліпідограми та генетичне тестування FISH-методом на наявність мікроделеції 15-ї хромосоми, також надані рекомендації щодо вигодовування. Отримані такі результати: глюкоза — 5,44 ммоль/л, Т3 вільний 8,5 пмоль/л (4,0–11,5), Т4 вільний 21,0 пмоль/л (8,8–25,0), тиреотропний гормон 4,7 мМе/л (0,4–5,0); на УЗД органів черевної порожнини без суттєвої структурної патології. Генетичне тестування не було одразу проведено, оскільки подібні тести не проводились у нашому регіоні на той час. Дитина знаходилась під наглядом педіатра, невролога, отримувала консультації генетика, ендокринолога, уролога, отримувала симптоматичне лікування, ЛФК, корекцію раціону харчування (строгий режим прийому їжі, обмеження легкозасвоюваних вуглеводів та жирів). У вересні 2014 року (у віці 1 рік 8 міс.) проведено каріотипування та діагностику мікроделеції 15-ї хромосоми FISH-методом: каріотип 46, XY.ish del(15)(15q11-15q13)(D15Z1+, D15S10-, PML+). Хлопчик знаходиться під наглядом профільних спеціалістів, регулярно проходить базові конт–рольні обстеження згідно з рекомендаціями (див. протокол № 55 від 03.02.09).
З віком спостерігається тенденція до підвищення апетиту, помірного пришвидшення набору ваги, що контролюється завдяки дотриманню дієти. Поступово проявляється помірне відставання у зрості (–2,39 стандартного відхилення та збільшення індексу маси тіла (18,9–2,0 стандартного відхилення). Моторний розвиток наближається до вікових показників, але спостерігається значне відставання в розвитку експресивного мовлення та помірне відставання в розумінні мовлення.
Висновки
Синдром Прадера — Віллі має характерні клінічні прояви, які в більшості пацієнтів можуть бути виявлені вже на першому півріччі життя, що дозволить провести своєчасну корекцію метаболічних і гормональних розладів та покращити прогноз для кожного окремого пацієнта. Для уточнення діагнозу з успіхом проводиться генетичне тестування, що на сьогодні стало доступне в більшості регіонів нашої країни. Відкритим залишається питання підтримуючої гормонотерапії СТГ, що може запобігати ряду ускладнень, характерних для синдрому, та покращити якість життя дітей із синдромом Прадера — Віллі в майбутньому.
Список литературы
1. Medical management for adults with Prader-Willi syndrome / Hauber M., Stratmann B., Hoedebeck-Stuntebeck N., Tschoepe D. // Metab. Syndr. Relat. Disord. — 2013 Dec. — 11(6). — 392-6.
2. Prader-Willi syndrome as a model of human hyperphagia / Tauber M., Diene G., Mimoun E., Çabal-Berthoumieu S., Mantoulan C., Molinas C., Muscatelli F., Salles J.P. // Front Horm. Res. — 2014. — 42. — 93-106.
3. Sarda P. Prader-Willi syndrome / P. Sarda // Soins Pediatr. Pueric. — 2013 Sep-Oct. — 274. — 20-3.
4. Rice L.J. Cognitive and behavioural aspects of Prader-Willi syndrome / Rice L.J., Einfeld S.L. // Curr. Opin. Psychiatry. — 2015 Mar. — 28(2). — 102-6.
5. Prenatal diagnosis of Prader-Willi syndrome and Angelman syndrome for fetuses with suspicious deletion of chromosomal region 15q11-q13 / Chang C.W., Hsu H.K., Kao C.C., Huang J.Y., Kuo P.L. // Int. J. Gynaecol. Obstet. — 2014 Apr. — 125(1). — 18-21.
6. Prader-Willi syndrome / Cassidy S.B., Schwartz S., Miller J.L., Driscoll D.J. // Genet Med. — 2012 Jan. — 14(1). — 10-26.
7. Cassidy S.B. Prader-Willi syndrome / Cassidy S.B., Driscoll D.J. // Eur. J. Hum. Genet. — 2009 Jan. — 17(1). — 3-13.
8. Cassidy S.B. Prader-Willi and Angelman syndromes: sister imprinted disorders / Cassidy S.B., Dykens E., Williams C.A. // Am. J. Med. Genet. — 2000. — 97(2). — 136-46.
9. Everman D.B. Genetics of childhood disorders: XII. Genomic imprinting: breaking the rules / D.B. Everman, S.B. Cassidy // J. Am. Acad. Child. Adolesc. Psychiatry. — 2000 Mar. — 39(3). — 386-9.
10. Growth hormone therapy, muscle thickness, and motor development in Prader-Willi syndrome: an RCT / Reus L., Pillen S., Pelzer B.J., van Alfen-van der Velden J.A., Hokken-Koelega A.C., Zwarts M., Otten B.J., Nijhuis-van der Sanden M.W. // Pediatrics. — 2014 Dec. — 134(6). — e1619-27.
11. Prader-Willi syndrome: A case report and a Chinese literature review / Zhu J., Cao Q., Zhang N., Zhao L. // Intractable Rare Dis. Res. — 2013 Nov. — 2(4). — 123-6.
12. Prader-Willi syndrome: a case report with atypical developmental features / Sewaybricker L.E., Guaragna-Filho G., Paula G.B., Andrade J.G., Tincani B.J., D’Souza-Li L., Lemos-Marini S.H., Maciel-Guerra A.T., Guerra-Júnior G. // J. Pediatr. Endocrinol. Metab. — 2014 Sep. — 27(9-10). — 983-8.
13. Sarda P. Prader-Willi syndrome / Sarda P. // Soins. Pediatr. Pueric. — 2013 Sep-Oct. — 274. — 20-3.
14. Синдромы Прадера — Вилли и Ангельмана: возможности молекулярно-цитогенетической и цитогенетической диагностики / Юров И.Ю., Ворсанова С.Г., Куринная О.С., Колотий А.Д., Демидова И.А., Кравец В.С., Юров Ю.Б. // Журнал неврологии и психиатрии им. C.C. Корсакова. — 2014. — Т. 114, № 1. — С. 49-53.
15. Особенности ожирения и метаболических нарушений при синдроме Прадера — Вилли у детей / Волеводз Н.Н., Богова Е.А., Немцова М.В., Ермакова М.А., Чернова Т.О., Сазонова Н.И. // Проблемы эндокринологии. — 2014. — Т. 60, № 1. — С. 24-31.
16. Тозлиян Е.В. Синдром Прадера — Вилли в практике педиатра // Практика педиатра. — 2014. — Т. 2. — С. 32-39.
17. Богова Е.А. Синдром Прадера — Вилли: новые возможности в лечении детей / Богова Е.А., Волеводз Н.Н. // Проблемы эндокринологии. — 2013. — Т. 59, № 4. — С. 33-40.
18. Гузева В.И. Клинические трудности диагностики синдрома Прадера — Вилли / Гузева В.И., Бессонова Л.Б., Сеель К.А. // Педиатр. — 2013. — Т. 4, № 2. — С. 81-84.