
Журнал «Здоровье ребенка» 4 (64) 2015
Вернуться к номеру
Механизм действия активированных кислородсодержащих метаболитов в респираторном тракте. Провоспалительное действие (часть 2)
Авторы: Абатуров А.Е. - ГУ «Днепропетровская медицинская академия Министерства здравоохранения Украины»; Волосовец А.П. - Национальный медицинский университет им. А.А. Богомольца, г. Киев
Рубрики: Педиатрия/Неонатология
Разделы: Справочник специалиста
Версия для печати
Резюме
В обзоре даны общие представления о механизмах провоспалительного действия активированных кислородсодержащих метаболитов.
В огляді подано загальні уявлення про механізми прозапальної дії активованих кисневмісних метаболітів.
The review presents general ideas about the mechanisms of proinflammatory action of activated oxygen-containing metabolites.
Ключевые слова
активированные кислородсодержащие метаболиты, легкие.
активовані кисневмісні метаболіти, легені.
activated oxygen-containing metabolites, lungs.
Статья опубликована на с. 124-130
Введение
Активированные кислородсодержащие метаболиты (АКМ) являются активными регуляторами воспалительного процесса, которые влияют на активность множества компонентов внутриклеточных сигнальных путей: AHR, AP-1, ATM, цAMФ, цAMФ-зависимой PKA, CDK5, c-myc, CREB, циклинов, FOXO, HIF-1α, JAK/STAT, JNK, MAPK, mTor, NF-κB, NFR2, PI3K/Akt, p38, p53, PKC, PPARγ, PTEN, PTPs/PTKs, SP1, WNT и других [29, 37].
Особую роль в активации внутриклеточных путей играют АКМ, продуцируемые в редоксосомах (редокс-активных эндосомах) в результате рецептор-ассоциированного возбуждения. Так, например, в состоянии покоя клетка экспрессирует NOX, анионные каналы и рецепторы IL-1R1 или TNFR1 на поверхности цитоплазматической мембраны. После взаимодействия IL-1β и TNF-β со специфическими для них рецепторами происходят образование рецептосомы и рекрутирование адаптерных молекул (MyD88 или TRADD). В последующем индуцируется эндоцитоз участка мембраны, в который вмонтированы IL-1R1 и компоненты NOX, удерживаемые рядом в составе липидного рафта за счет повышенной концентрации холестерина. В процессе эндоцитоза активное участие принимает кавеолин, «отшнуровывание» везикулы от плазмалеммы происходит при помощи динамина. В стенке сформированной таким образом эндосомы происходит перемещение субъединиц phoxсо сборкой НАДФН-оксидазы, которая начинает генерировать супероксид анион-радикал во внутреннее пространство, за счет чего эти везикулы получили название «редоксосомы» [25].
Супероксид анион-радикал, не обладая способностью самостоятельно проникать через мембрану эндосомы, накапливается и дисмутирует в перекись водорода, молекулы которой легко преодолевают мембранный барьер редоксосомы и проникают в цитоплазму клетки. Некоторая часть супероксид анион-радикала также попадает в цитоплазму клетки, но через хлоридные каналы (ClC-3, IClswell). Локализованная продукция перекиси водорода на поверхности редоксосомы приводит к передаче редокс-специфических либо рецептор-ассоциированных сигналов на нижерасположенные в сигнальном каскаде компоненты (IRAK/TRAF6 или TRAF2), что обусловливает активацию фактора транскрипции NF-κB. Накопление в цитоплазме перекиси водорода ведет к окислению протеина Rac1 на поверхности эндосомы/редоксосомы. И, как следствие процесса окисления, резко снижается уровень ассоциации между молекулами Rac1 и SOD1. Протеин Rac1 в отсутствие взаимосвязи с SOD1 быстро гидролизует ГТФ и становится неактивным, что обу–словливает прекращение NOX-ассоциированной продукции супероксид анион-радикала [25].
Среди множества редокс-модифицируемых протеинов, которые участвуют во внутриклеточной сигнализации, особое место занимают рецепторы, киназы, фосфатазы, факторы транскрипции и пероксиредоксины. Данные протеины содержат редокс-активные цистеиновые или метиониновые остатки, окисление которых изменяет активность возбуждения сигнальных путей (табл. 1) [30].
/125.jpg)
Редоксосомы также могут участвовать в функционировании антиген-представляющих клеток адаптивной иммунной системы. Учитывая, что окислительно-восстановительное состояние поздних эндосом в антиген-представляющих клетках регулируется IFN-γ-индуцибельными лизосомальными тиол-редуктазами (GILT), которые редуцируют дисульфидные связи эндосомальных протеинов в моноцитах, предполагают, что в ранних эндосомах тиолы данных протеинов окисляются во время NOX-опосредованных окислительных событий [30].
Провоспалительное действие АКМ
Провоспалительные эффекты АКМ обусловлены: 1) их способностью активировать TLR; 2) возбуждать МАРК-ассоциированные пути; 3) индукцировать активность ядерного фактора κB (NF-κB), факторов сигнальной трансдукции и активации транскрипции (STAT), активирующего протеина-1 (АР-1), фактора-1 раннего ростового ответа (EGR-1 — early growth response factor-1) и других факторов транскрипции [15, 27], способностью Н2О2 непосредственно активировать киназы IκB [9]; 4) влиять на эпигенетические механизмы активации провоспалительных генов [1]; 5); образовывать в результате взаимодействия с протеинами, липидами окисленные галогены, органические гидропероксиды ROOH, обладающие провоспалительным действием [2, 5].
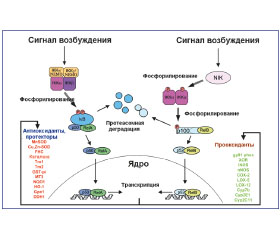
Показано, что АКМ могут как непосредственно активировать TLR2, TLR4 альвеолярных макрофагов, так и усиливать возбуждение их сигнальных путей, способствуя провоспалительной реакции (рис. 1) [19, 38, 39].
/127.jpg)
АКМ являются важнейшими модуляторами активности МАРК. Наиболее чувствительными к действию АКМ молекулами являются PTP (protein-tyrosine phosphatase — тирозиновая протеинфосфатаза), фосфорилирование которых в настоящее время признано в качестве основного регулирующего механизма внутриклеточной сигнализации [16, 26]. АКМ активируют рецепторные PTP и несколько нерецепторных тирозиновых протеинкиназ, принадлежащих к семейству Src и Janus киназ. Показано, что под влиянием АКМ повышается активность экстрацеллюлярной сигнал-регулируемой киназы (ERK), C-Jun N-терминальной киназы (JNK), p38-киназы, фосфатидилинозитол-3 киназы (PI3K), что приводит к активации факторов транскрипции –АР-1, ATF2, CBP, ELK-1 [7, 27].
По мнению John J. Haddad [10], изменения в структуре генной экспрессии клеток, происходящие под влиянием регулирующих факторов транскрипции, являются ведущими компонентами механизмов, которые определяют клеточные ответные реакции на флуктуации окислительно-восстановительного потенциала. АКМ изменяют активность многих факторов транскрипции (табл. 2) [29].
/126.jpg)
На протяжении более десяти лет известно, что NF-κB является кислород-сенситивным фактором транскрипции. NF-κB-ассоциированная активация продукции провоспалительных цитокинов, в том числе и IL-1β, нуждается в АКМ, в частности генерируемых НАДФ-оксидазой. АКМ-ассоциированная индукция фактора транскрипции NF-κB в клетках бронхопульмональной системы приводит к возбуждению генов, ответственных за синтез интерлейкинов (IL-1α, IL-1β, IL-2, IL-3, –IL-6, IL-8/CXCL8, IL-12), TNF-α, лимфотоксина-α, IFN-β, гранулоцитарно-макрофагального колониестимулирующего фактора (GM-CSF), α-цепей рецептора IL-2, острофазовых белков (сывороточного амилоидного A-протеина; компонентов В, С3, С4 системы комплемента, кислого α1-гликопротеина), молекул адгезии (межклеточной адгезивной молекулы-1 (ICAM-1/CD54), межклеточной адгезивной молекулы 2 (ICAM-2/CD102), адгезивной молекулы 1 сосудистого эндотелия (VCAM-1), адгезивной молекулы 1 клеток слизистых оболочек (MACAM-1), Е-селектина, иммунорегуляторных молекул (легких κ-цепей иммуноглобулинов, инвариантных цепей), α- и β-рецепторных цепей TCR T-клеток; продуктов локусов A, B, C HLA — антигенов класса I и локусов DR, DQ, DP HLA — антигенов класса II; β2-микроглобулина; ингибитора κB, p53; транспортера, ассоциированного с процессингом антигена, iNOS, COX2 [10, 22, 23]. Однако другими авторами представлены данные о том, что АКМ не индуцируют, а, окисляя Cys62 протеина p50, ингибируют активность связывания фактора транскрипции NF-κB с ДНК [6, 20]. Оксиданты могут специфично ингибировать активность NF-κB-пути в эпителиальных клетках респираторного тракта, S-убиквитинилируя цистеиновый остаток 179 (Cys179) IKKβ [24]. Показано, что антиоксидантная терапия, применяемая при лечении заболеваний легких, подавляет активность фактора NF-κB [10].
Фактор транскрипции NF-κB изменяет уровень экспрессии про- и антиоксидантных генов (рис. 2).
/128.jpg)
АКМ индуцируют активность митоген-активируемых протеинкиназ (МАРК) — ERK 1/2, JNK, p38, которые возбуждают протеины c-Jun, Jun B, Jun D, c-Fos, Fos B, Fra-1, Fra-2, являющихся членами семейства фактора транскрипции АР-1. Фактор транскрипции АР-1 играет основную роль в регуляции активности значительного количества генов, которые участвуют в воспалении и иммунном ответе. В частности, АР-1 индуцирует синтез кателицидина, сурфактантных белков A и D, секреторного белка клеток Клара, обладающих выраженной антимикробной активностью. При инфекционно-воспалительных заболеваниях органов дыхания AP-1 активирует транскрипцию генов матриксных металлопротеиназ — MMP-1, MMP-2, MMP-12; цитокинов — IL-4, IL-5, IL-10; интерферонов; адгезинов — ICAM-1/CD54, ICAM-2/CD102, –VCAM-1, E-селектина; хемоаттрактантов — IL-8/CXCL8, CXCL12/SDF-1 [18, 28].
АКМ-зависимой активации факторов транскрипции STAT предшествует индукция рецептор-ассоциированного семейства Janus-киназ (JAK1, JAK2, JAK3 и TYK2), которые фосфорилируют белки STAT, обусловливая их перемещение в ядро клетки, где они связываются с cis-элементами ДНК и возбуждают экспрессию генов, ответственных за синтез провоспалительных цитокинов, молекул адгезии, iNOS [11], а также супероксиддисмутазы, генов, регулирующих рост клетки. Возбуждение STAT, так же как и NF-κB, ингибируется биоантиоксидантами [4].
АКМ достоверно ускоряют процесс ацетилирования гистоновых белков, тем самым усиливая транскрипцию провоспалительных генов [33]. В экспериментальных работах было показано, что под влиянием АКМ в бронхиальных и альвеолярных эпителиоцитах, альвеолярных макрофагах респираторного тракта увеличивается экспрессия генов TNF-α, IL-1, IL-6, IL-8/CXCL8, ICAM-1/CD54, CCL3/MIP-1α, GM-CSF, iNOS, COX-2 [27].
Перекись водорода индуцирует активное и пассивное высвобождение HMGB1 из макрофагов и моноцитов дозозависимым способом. По всей вероятности, высвобождение HMGB1 связано с активацией МАРК- и CRM1-ассоциированных сигнальных путей [17]. Амфотерин (HMGB1) — негистоновый высококонсервативный хроматин-ассоциированный box1-белок группы протеинов высокой мобильности, который в физиологических условиях конститутивно экспрессируется и локализуется в ядре клетки, где принимает участие в регуляции транскрипции генов, ремодуляции хроматина и репарации ДНК [32].
Мыши с нокаутным геном HMGB1 погибают от выраженной гипогликемии в первые 24 часа жизни. В каждой клетке содержится приблизительно 106 молекул HMGB1. Протеин HMGB1 пассивно высвобождается при некротической (но не апоптотической) гибели клетки и активно секретируется макрофагами после индукции не только АКМ, но и IFN-γ, TNF-α и агонистами TLR3, TLR4 [8, 31]. Высвобожденный протеин HMGB1 взаимодействует по крайней мере с пятью различными мембранными рецепторами клеток иммунной системы: RAGE, TLR-2, TLR-4, триггерным рецептором, экспрессируемым на миелоидных клетках 1 (triggering receptor expressedon myeloidcells 1 — TREM1) и CD24, которые активируют MAPK, –NF-κB и PI3K/AKT сигнальные пути. HMGB1, непосредственно взаимодействуя с TLR2, TLR4, TLR9, индуцирует матурацию DC и вызывает активацию макрофагов, Т-лимфоцитов, эндотелиоцитов, обусловливая продукцию провоспалительных цитокинов (TNF-α, IL-1F1/IL-1α, IL-1F2/IL-1β, IL-6, IL-8/CXCL8) [13, 14, 21, 36]. Взаимодействие HMGB1 с TLR4 способствует увеличению экспрессии и представлению TLR2 на поверхности цитоплазматической мембраны альвеолярных макрофагов и эндотелиоцитов сосудов легких. Ассоциация HMGB1/TLR4 индуцирует TLR4-MyD88-IRAK4-сигнальный путь, что приводит к возбуждению p38 MAPK и Akt-пути, обусловливая индуцибельную активацию НАДФН-оксидазы, экспрессию ICAM-1, макрофагальную продукцию IL-23. В свою очередь, IL-23 через IL-17-G-CSF-опосредованный механизм вызывает высвобождение полиморфноядерных лейкоцитов из костного мозга в периферическое русло крови. Однако были представлены экспериментальные данные и о том, что HMGB1 непосредственно не вызывает активацию TLR [12, 35].
Однако окисление аминокислотного остатка Cys106 молекулы HMGB1 является достаточным для того, чтобы заблокировать иммуногенное действие данного протеина на дендритные клетки [34].
Список литературы
1. Afanas’ev I. New nucleophilic mechanisms of ros-dependent epigenetic modifications: comparison of aging and cancer // Aging Dis. — 2013, Oct 21. — 5(1). — 52-62. — doi: 10.14336/AD.2014.050052.
2. Biomarkers of Oxidative Damage in Human Disease / I. Dalle-Donne, R. Rossi, R. Colombo, D. Giustarini, A. Milzani // Clin. Chem. — 2006 Apr. — 52(4). — 601-23. — doi: 10.1373/clinchem.2005.061408.
3. Brigelius-Floh R., Floh L. Basic principles and emerging concepts in the redox control of transcription factors // Antioxid. Redox. Signal. — 2011, Oct 15. — 15(8). — 2335-81. — doi: 10.1089/ars.2010.3534.
4. Comhair S.A., Erzurum S.C. Antioxidant responses to oxi–dant-mediated lung diseases // Am. J. Physiol. Lung. Cell. Mol. Physiol. — 2002 Aug. — 283(2). — L246-55. — doi: 10.1152/ajplung.00491.2001.
5. Davies M.J. The oxidative environment and protein damage // Biochim. Biophys. Acta. — 2005, Jan 17. — 1703(2). — 93-109.
6. Evidence that reactive oxygen species do not mediate NF-kB activation / M. Hayakawa, H. Miyashita, I. Sakamoto, M. Kitagawa, H. Tanaka, H. Yasuda, M. Karin, K. Kikugawa // EMBO J. — 2003, Jul 1. — 22(13). — 3356-66. — doi: 10.1093/emboj/cdg332.
7. Free radicals and antioxidants in normal physiological functions and human disease / M. Valko, D. Leibfritz, J. Moncol, M.T. Cronin, M. Mazur, J. Telser // Int. J. Biochem. Cell. Biol. — 2007. — 39(1). — 44-84.
8. Gauley J., Pisetsky D.S. The translocation of HMGB1 du–ring cell activation and cell death // Autoimmunity. — 2009 May. — 42(4). — 299-301.
9. H2O2 in the induction of NF-κB-dependent selective gene expression / L. Cyrne, V. Oliveira-Marques, H.S. Marinho, F. Antunes // Methods Enzymol. — 2013. — 528. — 173-88. — doi: 10.1016/B978-0-12-405881-1.00010-0.
10. Haddad J.J. Science review: Redox and oxygen-sensitive transcription factors in the regulation of oxidant-mediated lung injury: role for nuclear factor-κB // Crit. Care. — 2002 Dec. — 6(6). — 481-90. — doi: 10.1186/cc1839.
11. Harrison D.A. The Jak/STAT pathway // Cold Spring Harb. Perspect Biol. — 2012, Mar 1. — 4(3). — pii: a011205. — doi: 10.1101/cshperspect.a011205.
12. High mobility group box 1 protein binding to lipopolysaccharide facilitates transfer of lipopolysaccharide to CD14 and enhances lipopolysaccharide-mediated TNF-α production in human monocytes / J.H. Youn, Y.J.Oh, E.S. Kim, J.E. Choi, J.S. Shin // J. Immunol. — 2008, Apr 1. — 180(7). — 5067-74. — doi: 10.4049/jimmunol.180.7.5067.
13. HMGB1 and RAGE in inflammation and cancer / G.P. Sims, D.C. Rowe, S.T. Rietdijk, R. Herbst, A.J. Coyle // Ann. Rev. Immunol. — 2010. — 28. — 367-88. — doi: 10.1146/annurev.immunol.021908.132603.
14. HMGB1: endogenous danger signaling / J.R. Klune, R. Dhupar, J. Cardinal, T.R. Billiar, A. Tsung // Mol. Med. — 2008 Jul — Aug. — 14(7–8). — 476-84. — doi: 10.2119/2008-00034.Klune.
15. Hsu H.-Y., Wen M.-H. Lipopolysaccharide-mediated Reactive Oxygen Species and Signal Transduction in the Regulation of Interleukin-1 Gene Expression // J. Biol. Chem. — 2002, Jun 21. — 277(25). — 22131-9.
16. Hunter T. Tyrosine phosphorylation: thirty years and coun–ting // Curr. Opin. Cell. Biol. — 2009 Apr. — 21(2). — 140-6. — doi: 10.1016/j.ceb.2009.01.028.
17. Hydrogen peroxide in inflammation: messenger, guide, and assassin / C. Wittmann, P. Chockley, S.K. Singh, L. Pase, G.J. Lieschke, C. Grabher // Adv. Hematol. — 2012. — 2012. — 541471. — doi: 10.1155/2012/541471.
18. Karamouzis V., Konstantinopoulos P.A., Papavassiliou A.G. The Activator Protein-1 Transcription Factor in Respiratory Epithelium Carcinogenesis // Mol. Cancer. Res. — 2007 Feb. — 5(2). — 109-20. — doi: 10.1158/1541-7786.MCR-06-0311.
19. Karki R., Igwe O.J. Toll-like receptor 4-mediated nuclear factor kappa B activation is essential for sensing exogenous oxidants to propagate and maintain oxidative/nitrosative cellular stress // PLoS One. — 2013, Sep 18. — 8(9). — e73840. — doi: 10.1371/journal.pone.0073840.
20. Krause K.-H. Tissue Distribution and Putative Physiological Function of NOX Family NADPH Oxidases // Jpn J. Infect. Dis. — 2004 Oct. — 57(5). — S28-9.
21. Lee K.-M., Seong S.-Y. Partial role of TLR4 as a receptor responding to damage-associated molecular pattern // Immunol. Lett. — 2009, Jun 30. — 125(1). — 31-9. — doi: 10.1016/j.imlet.2009.05.006.
22. Morgan M.J., Liu Z.G. Crosstalk of reactive oxygen species and NF-κB signaling // Cell. Res. — 2011 Jan. — 21(1). — 103-15. — doi: 10.1038/cr.2010.178.
23. Nordberg J., Arner E.S. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system // Free Radic. Biol. Med. — 2001, Dec 1. — 31(11). — 1287-312. — doi: 10.1016/S0891-5849(01)00724-9.
24. Nuclear factor kappaB, airway epithelium, and asthma: avenues for redox control / Y.M. Janssen-Heininger, M.E. Poynter, S.W. Aesif, C. Pantano, J.L. Ather, N.L. Reynaert, K. Ckless, V. Anathy, J. van der Velden, C.G. Irvin, A. van der Vliet // Proc. Am. Thorac. Soc. — 2009, May 1. — 6(3). — 249-55. — doi: 10.1513/pats.200806-054RM.
25. Oakley F.D., Abbott D., Li Q., Engelhardt J.F. Signaling components of redox active endosomes: the redoxosomes // Antioxid. Redox Signal. — 2009 Jun. — 11(6). — 1313-33. — doi: 10.1089/ARS.2008.2363.
26. Protein tyrosine phosphatase SHP-2 is positively involved in platelet-derived growth factor-signaling in vascular neointima formation via the reactive oxygen species-related pathway / K.J. Won, H.M. Lee, C.K. Lee, H.Y. Lin, H. Na, K.W. Lim, H.Y. Roh, S. Sim, H. Song, W.S. Choi, S.H. Lee, B. Kim // J. Pharmacol. Sci. — 2011. — 115(2). — 164-75 // http://dx.doi.org/10.1254/jphs.10250FP
27. Rahman I. Oxidative Stress, Chromatin Remodeling and Gene Transcription in Inflammation and Chronic Lung Diseases // J. Biochem. Mol. Biol. — 2003, Jan 31. — 36(1). — 95-109.
28. Rahman I., MacNee W. Oxidative stress and regulation of glutathione in lung inflammation // Eur. Respir. J. — 2000 Sep. — 16(3). — 534-54.
29. Reuter S., Gupta S.C., Chaturvedi M.M., Aggarwal B.B. Oxidative stress, inflammation, and cancer: how are they linked? // Free Radic. Biol. Med. — 2010, Dec 1. — 49(11). — 1603-16. — doi: 10.1016/j.freeradbiomed.2010.09.006.
30. Spencer N.Y., Engelhardt J.F. The basic biology of redoxosomes in cytokine-mediated signal transduction and implications for disease-specific therapies // Biochemistry. — 2014, Mar 18. — 53(10). — 1551-64. — doi: 10.1021/bi401719r.
31. Srikrishna G.H., Freeze H. Endogenous Damage-Associated Molecular Pattern Molecules at the Crossroads of Inflammation and Cancer // Neoplasia. — 2009 Jul. — 11(7). — 615-28. — PMCID: PMC2697348.
32. Štros M. HMGB proteins: Interactions with DNA and chromatin // Biochim. Biophys. Acta. — 2010 Jan — Feb. — 1799(1–2). — 101-13. — doi: 10.1016/j.bbagrm.2009.09.008.
33. Sundar I.K., Caito S., Yao H., Rahman I. Oxidative stress, thiol redox signaling methods in epigenetics // Methods Enzymol. — 2010. — 474. — 213-44. — doi: 10.1016/S0076-6879(10)74013-1.
34. Tang D., Kang R., Zeh H.J. 3rd, Lotze M.T. High-mobility group box 1, oxidative stress, and disease // Antioxid. Redox Signal. — 2011, Apr 1. — 14(7). — 1315-35. — doi: 10.1089/ars.2010.3356.
35. The alarmin HMGB1 acts in synergy with endogenous and exogenous danger signals to promote inflammation / H.S. Hreggvidsdottir, T. Ostberg, H. Wahamaa, H. Schierbeck, A.C. Aveberger, L. Klevenvall, K. Palmblad, L. Ottosson, U. Andersson, H.E. Harris // J. Leukoc. Biol. — 2009 Sep. — 86(3). — 655-62. — doi: 10.1189/jlb.0908548.
36. Tsung A., Tohme S., Billiar T.R. High-mobility group box-1 in sterile inflammation // J. Intern. Med. — 2014 Nov. — 276(5). — 425-43. — doi: 10.1111/joim.12276.
37. Wrzaczek M., Broschе́ M., Kangasjärvi J. ROS signa–ling loops — production, perception, regulation // Curr. Opin. Plant. Biol. — 2013 Oct. — 16(5). — 575-82. — doi: 10.1016/j.pbi.2013.07.002.
38. Xiang M., Fan J. Pattern recognition receptor-dependent mechanisms of acute lung injury // Mol. Med. — 2010 Jan — Feb. — 16(1–2). — 69-82. — doi: 10.2119/molmed.2009.00097.
39. Xiang M., Fan J., Fan J. Association of Toll-like receptor signaling and reactive oxygen species: a potential therapeutic target for posttrauma acute lung injury // Mediators Inflamm. — 2010. — pii: 916425. — doi: 10.1155/2010/916425.