
Журнал «Здоровье ребенка» 4 (64) 2015
Вернуться к номеру
The Prader-Willie syndrome in practice of pediatrician, modern approaches to diagnostics and treatment
Авторы: Khimenko T.M., Adachovska A.O. - Odessa National Medical University
Рубрики: Педиатрия/Неонатология
Разделы: Справочник специалиста
Версия для печати
Prader-Willie syndrome, diagnostics and treatment, growth hormone.
Prader-Willi syndrome (PWS) is thought to be one of the more common disorders seen for genetic services, and the most frequent recognized genetic form of childhood obesity and affects an estimated 350,000 – 400,000 people worldwide [2]. According to The Global Prader-Willi syndrome Registry this complex multisystem genetic disorder occurs in approximately one out of every 15,000 births. PWS is caused by abnormalities of the imprinted region of proximal 15q and results from absence of the normally active paternally inherited genes in this region at chromosome 15(15q11-q13). It is of interest that a clinically very different disorder, Angelman syndrome, is the result of an oppositely imprinted gene in the same region of chromosome 15. Although this syndrome includes specific features many children with PWS were not diagnosed until rapid weight gain leading to obesity was evident and the presence of specific learning/behavioral problems was observed. In according to Butler the average age at diagnosis for PWS was greater than 6 years of age. The manifestations of PWS are related to functional hypothalamic deficiency, the clinical appearance in infants differs considerably from that in childhood and adulthood [1]. Early natural history of PWS can be divided into two distinct clinical stages. The first stage occurs during the neonatal and early infancy periods and is characterized by varying degrees of hypotonia, a week cry, development delay, temperature instability, a poor suck reflex, sticky saliva, feeding difficulties sometimes requiring gastrostomy or stomach tube placement, hypogonadism at birth as genital hypoplasia. Failure-to-thrive is noted during this first stage. The hypotonia is thought to be central in origin, non-progressive, and on the average begins to improve between 8 and 11 months of age. The second stage usually begins around 2 years of age and is characterized by continued developmental delay or psychomotor retardation and onset of hyperphagia leading to obesity. Other features noted during the second stage may include speech articulation problems, foraging for food, rumination, unmotivated sleepiness, physical inactivity, decreased pain sensitivity, skin picking and other forms of self-injurious behavior, prolonged periods of hypothermia, strabismus, hypopigmentation, scoliosis, obstructive sleep apnea, and abnormal oral pathology (enamel hypoplasia, dental caries, malocclusion and decreased saliva). Temper tantrums, depression, stubbornness, obsessive compulsivity, and sudden acts of violence of varying degrees may be observed during this stage. Poor peer interactions, immaturity, and inappropriate social behavior may also occur during this time. Phenotype can include narrow bifrontal diameter, almond shaped palpebral fissures, narrow nasal bridge, and downturned mouth with a thin upper lip, hypopigmentation of skin, hair, eye colour, strabism are either present from birth or evolve over time. Small narrow hands with a straight ulnar border and sometimes tapering fingers, and short, often broad feet, are usually present by the age of 10, with an average adult foot length of 20,3 cm in females, 22.3 cm in males. Holm, Cassidy, Butler, and others developed consensual diagnostic criteria to assist in the diagnosis for PWS using major, minor and supportive features and established a scoring system for patients presenting with features seen in the syndrome. To confirm the diagnosis of PWS should be done genetic testing. This testing includes chromosomal or microarray analysis and assessment for methylation patterns in PWS region. Fluorescent in situ hybridization (FISH) can be used to confirm prenatal diagnosis when a deletion in the 15q region is suspected after chorionic villus sampling or amniocentesis [3, 1, 7].
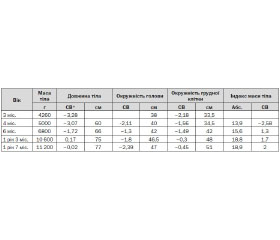
Symptomatic treatment significantly affects the quality and length of life of people with PWS. According to The order of Ministry of Health of Ukraine № 55 to provide medical care to children with PWS from 29.08.2006 therapy of this syndrome should be directed to the 6 main units: initial management of hypotonia or poor feeding, evaluation for hypogonadism or hypopituitarism, management of obesity, treatment of growth delay, therapy for behavioral issues, treatment of complications and other manifestations of PWS (diabetes mellitus, sleep apnea, syndrome Pickwick, monitoring of scoliosis, correction of strabismus hypertension, thrombophlebitis, chronic leg edema and others) [8]. Obesity is the major cause of morbidity and mortality in PWS and longevity may be nearly normal if it is avoided. To prevent obesity recommended balanced hipocaloric diet (1000 calories with the addition of vitamins and calcium) from early school age under the supervision of a nutritionist. Treatment with growth hormone decreases body weight, corrects osteopenia, prevents development of scoliosis and correct change of behavior. On June 20, 2000, the US Food and Drug Administration (FDA) approved the use of growth hormone in children with genetically confirmed PWS and evidence of growth failure [7]. In Ukraine growth hormone drug is recommended to take a daily subcutaneously or 6 days a week. The total weekly dose is – 0,15-0,3 mg/kg; from further correction of the effective dose [8].
Conclusion. Prader-Willie syndrome has typical clinical manifestations, which can be seen during first year of life, that enables us to carry out the correction of metabolic and hormone disorders and to improve quality of life. Genetic test is held to clarify the diagnosis. There is still open question concerning supportive hormone therapy which can correct a phenotype, growth and proportions of a patient. Long-term application of recombinant growth hormone may reduce certain complications and raise the quality of patient’s life in future.