
Журнал «Здоровье ребенка» 4 (64) 2015
Вернуться к номеру
Orphan hereditary syndromes in childish endocrinologist
Авторы: M.O.Riznichuk, V.P.Pishak - Bukovinian State Medical University
Рубрики: Педиатрия/Неонатология
Разделы: Справочник специалиста
Версия для печати
orphan hereditary syndromes, gigantism, children, genes.
Summary. Orphan syndromes is a group of rare diseases, with a rate of 10 cases per 100 000 population. Considerable attention of physicians of different profiles requires orphan syndromes with lesions of the endocrine system. Phenotypic these syndromes begin to appear from the birth or they appear in the childhood. In this article is given a short phenotypic and genetic characteristics of certain orphan endocrine syndromes, accompanied with gigantism.
Perlman syndrome (MIM 267000). Phenotypic feature of this syndrome are ascites without edema of the fetus and polyhydramnios.
Sick children are characterized by a large mass at birth, muscular hypotonia, organomegaly, typical facial dysmorphism, renal anomalies, agenesis of the corpus callosum, delayed psychomotor development, hyperinsulinemia and high infant mortality.
Perlman syndrome causes a high risk of Wilms tumor. This pathology is caused by homozygous or heterozygous mutations in the gene DIS3L2 (MIM 614184) on chromosome 2q37. Inheritance - autosomal recessive.
Proteus syndrome (MIM 176920). The cause of the syndrome is spontaneous variation of the gene during embryonic development. The severity of disease depends on the period of embryogenesis, when actually, there was a genetic mutation that causes to the formation of at least three sub-cells - normal, atrophic and hypertrophic.
Proteus syndrome is accompanied by the growth of skin, bones, muscles, fat, blood and lymph vessels. Phenotypic manifestations of Proteus syndrome are found in the first 2 years of life. Strabismus, exophthalmos, myopia, progeny, varicose veins, skin growths on the soles are diagnosed.
With age, there is too rapid tumor growth. The syndrome is associated with mutations in the gene AKT1 (MIM 164730) on chromosome 14q32.3.
Inheritance - autosomal dominant.
Sotos syndrome 1 (MIM 117550). The syndrome of children's overgrowth, is characterized by excessive growth, certain cranio-facial features, mental retardation and increased bone age.
In the absence of one or more of the four criteria (height more than 97 percentile and head circumference more than 97 percentile, bone age > 90th percentile, mental retardation) diagnosis is unlikely. The disease manifests from birth. Typical cranio-facial dysmorphiy: macrodolihocephaliya, protruding frontal mounds, coarse facial features with hipertelorysmus, strabismus, protruding lower jaw (prognathism), macroglossia and gothic palate. Bone changes manifested by following features: large hands and feet, kifo-scoliosis, webbing foot. On the internal organs is sometimes visceromehaliya. The final height is on the top of percentile. Abnormal dermatoglyphics, normal level of hormone of growth and high levels of valine, isoleucine and leucine levels.
It is called by heterozygous mutation in the gene NSD1 (MIM 606681) in the region 5q35.
Inheritance - autosomal dominant, most cases - sporadic. Life is not changed. It has good prediction, treatment is not carried out.
Sotos syndrome 2 (MIM 614753). It is called by heterozygous mutation in the gene NFIX (MIM 164005) on chromosome 19p13.3. There is intense postnatal linear growth, macrocephaly, bone age advance, long narrow face, high forehead, scoliosis. Unusual behavior is characterized by development of anxiety and mental retardation.
Berardinelli syndrome (MIM 269700). Molecular and genetic analysis prove the existence of 4 types of congenital generalized lipodystrophy. Type 1 is caused by mutation in the gene AGPAT2, encoding 1-acyl-glycerol-O-acetyltransferase 2. Gene is located in the locus 9q34. Type 2 of syndrome develops due to mutation in the gene locus BSCα2 in 11q13. Gene product is protein seypin BSCα2 contains 398 amino acids involved in the differentiation and functioning of adipocytes. Molecular-genetic basis of Berardinelli-Seypa syndrome type 3 is mutation in the gene CAV1 (locus 7q31) in participation of which is synthesized protein caveolin 1 – an integral part of the membrane vesicles. Congenital generalized lipodystrophy, type 4 is caused by mutations in a gene PTRF located on chromosome 17 and encodes a protein PTRF (polymerase and transcript release factor).
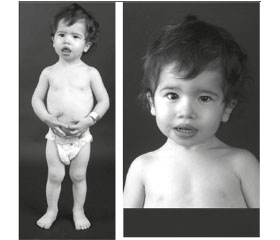
Patient's inherent rapid growth, increased appetite and a special appearance. Lack of subcutaneous adipose tissue in combination with increased muscle tissue forms in such patients expressed muscle relief. Other features: great chin, large hands and feet, hirsutism, acantosis nigrigans. Female patients has enlarged clitoris and males - increase external genitalia.
By this announcement we have attempted to draw the attention of practitioners to clearly outline group of orphan diseases. Obviously, there is a need for a common approach to their definition, classification and diagnostic criteria.