Статья опубликована на с. 136-140
Введение
Активированные кислородсодержащие метаболиты (АКМ) оказывают разностороннее действие в респираторном тракте, в том числе вызывают противовоспалительный эффект, регулируют процессы апоптоза и выживания клеток.
Противовоспалительное действие АКМ
В основе противовоспалительного действия АКМ, по мнению C. Milla и соавт. [25], лежит их способность добиваться снижения уровня продукции провоспалительных цитокинов, индуцируя апоптоз Т-лимфоцитов. Также связанная с действием АКМ активация факторов транскрипции приводит к продукции некоторых цитопротекторных протеинов.
АКМ способствуют возбуждению факторов транскрипции ядерного фактора-2, подобного эритроидному деривату-2 NRF2 (NFE2L2 — nuclear factor (erythroid-derived 2)-like 2), NF-κB, АР-1, ядерного протеина IL-6 (NF-IL-6 — nuclearprotein IL-6) [29].
Индукция АКМ фактора транскрипции NRF2 приводит к возбуждению транскрипции более 200 генов, ответственных за синтез ферментов детоксикации и протеинов с антиоксидативной активностью — супероксиддисмутаз, каталазы, глутатион-S-трансферазы (GST), глутатионпероксидазы (GPx), гемоксигеназы-1 (HO-1) и других [21, 22].
Возбуждение фактора транскрипции NF-κB обу–словливает синтез MnSOD, GPx; факторов транскрипции STAT — HO-1 [6, 9].
В эндотелиальных клетках легочных сосудов АКМ через возбуждение фактора транскрипции NF-IL-6 активируют транскрипцию гена IL-6. Известно, что IL-6 является одним из основных индукторов продукции большинства острофазовых белков. Однако ZhouXing и соавт. [34] показали, что IL-6 играет важную противовоспалительную роль в развитии местной и системной острой воспалительной реакции. IL-6 ингибирует продукцию провоспалительных, но не противовоспалительных цитокинов, и эта противовоспалительная активность IL-6 не может быть компенсирована действием –IL-10.
Влияние АКМ на апоптоз и клеточный цикл
/137-1.jpg)
АКМ участвуют в регуляции апоптоза клеток респираторного тракта. Показано, что альвеолоциты I и II типа отличаются высокой чувствительностью к проапоптотическому действию АКМ [15, 16]. АКМ индуцируют путь рецепторов смерти и митохондриальный путь апоптоза (рис. 1). Перекись водорода также индуцирует активность цитоплазматического транспорта Fas на поверхность мембран эпителиальных клеток респираторного тракта, увеличивая вероятность апоптотической гибели клетки [18, 19]. Высокий уровень АКМ способствует активации ASK1/JNK-сигнального пути, возбуждение которого индуцирует апоптоз клеток. Необходимо отметить, что только пролонгированная активация JNK приводит к развитию апоптоза. АКМ опосредованно через ERK 1/2 способны активировать каспазу-3 и усиливать экспрессию церамидов и проапоптотических протеинов BAX и BAK, которые, взаимодействуя, создают порообразные структуры в наружной мембране митохондрии [14]. АКМ, вызывая пермеабилизацию митохондриальной наружной мембраны, индуцируют высвобождение в цитоплазму клетки цитохрома C, проапоптотических протеинов: Smac/Diablo и Omi/HtrA2В, AIF и endoG. В цитоплазме клетки цито–хром C и АТР/дезоксиАТР взаимодействуют с адаптерной молекулой — фактором, активирующим апоптотические протеазы APAF-1 (apoptotic protease activating factor), вызывая олигомеризацию APAF-1.
Олигомеризация APAF-1 обусловливает экспонирование домена CARD, что ведет к CARD-CARD взаимодействию APAF-1 и прокаспазы-9, организующему уникальную молекулярную конструкцию — апоптосому. Молекула APAF-1 в апоптосоме играет роль платформы, на которой происходят накопление и аутоактивация прокаспазы-9. В последующем активная каспаза-9 одновременно активирует каспазу-7 и каспазу-3. Каспаза-3, в свою очередь, возбуждает фактор фрагментации ДНК (DFF) и каспазо-активируемую дезоксирибонуклеазу (CAD), которая, вызывая межнуклеосомальные разрывы хроматина, нарушает целостность ДНК. Протеины Smac/Diablo и Omi/HtrA2В подавляют в цитоплазме клетки активность ингибиторов апоптогенных протеинов (IAP) — XIAR, cIAP1, cIAP2, сурвивина, аполлона. Белки AIF и endoG индуцируют каспазонезависимый апоптоз клеток. В то же время АКМ могут подавлять активность каспаз. В частности, Н2О2, окисляя цистеиновый остаток в каталитическом центре молекулы каспазы, обратимо инактивирует каспазу-3 и каспазу-8 [3, 4, 7, 28]. H2O2-индуцированная инактивация каспазы-9 опосредована ионом железа и также связана с окислением цистеинового остатка ее молекулы [2].
Однако основным механизмом развития H2O2-индуцированного апоптоза считают усиление экспрессии протеина p53. Протеин p53 вызывает продукцию p53-индуцированного протеина со смертельным доменом (DD), который через DD/DD-взаимодействие активирует адаптерный протеин RAIDD, содержащий домены DD и RIPK1. Протеин RAIDD рекрутирует прокаспазу-2, что обусловливает формирование PIDDосомы и активацию прокаспазы-2 [13, 17].
Потеря эпителиальных клеток за счет апоптоза — характерная особенность патогенеза острых респираторных инфекций, бронхиальной астмы и хронической обструктивной болезни легких. Показано, что эпителиоциты респираторного тракта у больных бронхиальной астмой высокочувствительны к апоптотическому действию H2O2 [18].
В эпителиальных клетках бронхов и альвеол уже через 48 часов после воздействия АКМ повышается экспрессия ингибитора циклинзависимых киназ p21CIP1, который играет активную роль в подавлении перехода клетки из фазы G1 в G1-S-фазу клеточного цикла [27].
Влияние АКМ на процессы репарации и выживание клеток
АКМ активируют макрофагальную продукцию трансформирующего фактора роста β1 (TGF-β1), фактора роста гепатоцитов, участвующих в репарации тканей респираторного тракта. IL-6, продукция которого индуцируется АКМ, предупреждает поражение легочной ткани, возбуждая синтез антиапоптотических протеинов bcl-2 и тканевого ингибитора металлопротеиназы-1 (TIMP-1) [15].
Активация АКМ фактора транскрипции STAT3 оказывает эпителиопротекторное действие в респираторном тракте, ингибируя продукцию матриксных металлопротеиназ MMP-9 и MMP-12 нейтрофилами [23]. Необходимо отметить, что IL-2 и IFN-κ, синтез которых индуцируется АКМ, также усиливают процессы репарации альвеолярного эпителия [20].
АКМ являются триггером для негипоксического возбуждения процессов транскрипции генов, ассоциированных с гипоксией, продукты которых обладают цитопротекторным действием. Активация этих генов сопровождается продукцией индуцибельного гипо–ксией фактора 1α (HIF-1α), протеина группы высокой мобильности бокс 1 (HMGB1), EGR-1, NF-IL-6 [10]. Предполагается, что в ответ на гипоксию возбуждается 1–1,5 % генов всего генома человека [8].
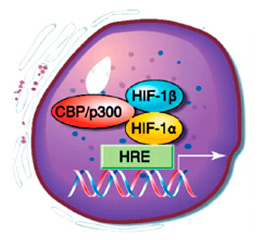
Молекула HIF-1 представляет гетеродимер семейства факторов транскрипции и состоит из двух субъединиц — индуцибельного гипоксией фактора HIF-1α и ядерного транслятора HIF-1β [33]. В условиях нормоксии под действием убиквитин-протеасомной системы HIF-1α быстро деградирует на фоне сохранения конститутивной экспрессии ядерного транслятора HIF-1β. Для убиквитинирования HIF-1α необходимо гидроксилирование двух пролиновых остатков (Pro402 и Pro564) его молекулы. Гидроксилирование HIF-1α осуществляется белками пролилгидроксилазного домена (PHD). Гидроксилированная молекула HIF-1α взаимодействует с протеином Von Hipple-Lindau (VHL) и подвергается убиквитинированию. Из известных четырех изоформ PHD молекулу HIF-1α могут гидроксилировать только PHD1–3, и основной изоформой, определяющей уровень гидро–ксилирования HIF-1α, является PHD2. При пониженной концентрации О2 снижается активность PHD, что приводит к недостаточному гидроксилированию пролиновых остатков и стабилизации молекулы HIF-1α [32]. Также развитие гипоксии сопровождается усилением синтеза шаперонов HSP70 и HSP90, которые защищают HIF-1α от убиквитин-протеасомной деградации и способствуют его кумуляции [35]. По достижении определенной концентрации HIF-1α перемещается в ядро клетки и, связываясь с cis-элементами промоторов определенных кислород-чувствительных генов, активирует или супрессирует их активность [10]. Активные радикалы кислорода — O2–•, OH• дифференцированно, в зависимости от pO2, влияют на экспрессию генов HIF-1α и ускоряют перемещение фактора транскрипции HIF-1α в ядро клетки [10, 11]. В настоящее время идентифицировано более 100 HIF-активируемых генов, продукты которых участвуют в эритропоэзе и обмене железа — эритропоэтин, трансферрин, трансферриновый рецептор, церулоплазмин; ангиогенезе — TGF-β3, сосудистый эндотелиальный фактор роста (VEGF), EG-VEGF, ММР-2, катепсина D; регуляции сосудистого тонуса — нитрооксидсинтаза-2 (iNOS), эндотелин-1, адреномедуллин, α1в-адренорецептор; регуляции метаболизма глюкозы — аденилаткиназа-3, альдолаза-А, -С, карбоангидраза-9, энолаза-1, транспортеры 1 и 3 глюкозы (GLUT1, GLUT3), гексокиназа-1 и -2, лактатдегидрогеназа-А, пируваткиназа-М, фосфофруктокиназа L, активируя анаэробное дыхание; регуляции клеточной пролиферации и выживания — IGF2, TGF-α, адреномедуллин; регуляции апоптоза — протеины BNip3, Nix (рис. 2) [12, 26, 30].
/138-1.jpg)
HIF-1α усиливает как процессы пролиферации, инициируя синтез пропролиферативных белков (IGF-2, IGF-BP-1, -3, TGF-β3), так и процессы апо–птоза, вызывая продукцию проапоптотических белков (DEC-1, Bcl2, NIX) [31].
Ответ на гипоксию тесно связан с иммунными реакциями, ассоциированными с NF-κB сигнальными путями. Активация фактора транскрипции NF-κB способствует экспрессии HIF-1α в макрофагах, нейтрофилах и эндотелиоцитах (рис. 3) [24].
Заключение
Течение инфекционно-воспалительных заболеваний органов дыхания сопровождается значительным увеличением концентрации активированных кислородсодержащих метаболитов как во внутритканевом пространстве, так и в бронхоальвеолярной жидкости. Возбуждение эпителиоцитов, альвеолярных макрофагов и нейтрофилов, характерное для инфекционно-воспалительных заболеваний респираторного тракта, сопровождается индукцией НАДФН-оксидазы. Основным продуктом функционирования НАДФН-оксидазы является супероксидный анион-радикал, который обладает бактерицидным действием и представляет важнейший компонент неспецифической противоинфекционной защиты человеческого организма. Киллинг микроорганизмов осуществляется за счет непосредственного окисления молекулярных структур инфекционных агентов или опосредованно через активацию нейтрофильных протеаз супероксидным анионом радикалом и перекисью водорода. Активированные кислородсодержащие метаболиты локально, с учетом короткой продолжительности их существования, регулируют через определенные факторы транскрипции экспрессию множества генов, участвующих в пролиферации, цитодифференцировке, регуляции апоптоза, выживания клеток и процесса воспаления.
Список литературы
1. Altieri D.C. Survivin and IAP proteins in cell-death mechanisms/ Оchem J. — 2010, Sep 1. — 430(2). — 199-205. — doi: 10.1042/BJ20100814.
2. Barbouti A. Hydrogen peroxide inhibits caspase-dependent apoptosis by inactivating procaspase-9 in an iron-dependent manner / A. Barbouti, C. Amorgianiotis, E. Kolettas, P. Kanavaros, D. Galaris // Free Radic. Biol. Med. — 2007, Nov 15. — 43(10). — 1377-87. — doi:10.1016/j.freeradbiomed.2007.06.020.
3. Bratton S.B., Salvesen G.S. Regulation of the Apaf-1-caspase-9 apoptosome // J. Cell. Sci. — 2010, Oct 1. — 123(Pt 19). — 3209-14. — doi: 10.1242/jcs.073643.
4. Budinger G.R. Epithelial cell death is an important contributor to oxidant-mediated acute lung injury / G.R. Budinger, G.M. Mutlu, D. Urich, S. Soberanes, L.J. Buccellato, K. Hawkins, S.E. Chiarella, K.A. Radigan, J. Eisenbart, H. Agrawal, S. Berkelhamer, S. Hekimi, J. Zhang, H. Perlman, P.T. Schumacker, M. Jain, N.S. Chandel // Am. J. Respir. Crit. Care Med. — 2011, Apr 15. — 183(8). — 1043-54. — doi: 10.1164/rccm.201002-0181OC.
5. Circu M.L., Aw T.Y. Reactive oxygen species, cellular redox systems, and apoptosis / Free Radic. Biol. Med. — 2010, Mar 15. — 48(6). — 749-62. — doi: 10.1016/j.freeradbiomed.2009.12.022.
6. Clark R.A., Epperson T.K. Anthony J. Mechanisms of Activation of NADPH Oxidases // Jpn J. Infect. Dis. — 2004 Oct. — 57(5). — S22-3. — PMID: 15507761.
7. Damgaard R.B., Gyrd-Hansen M. Inhibitor of apoptosis (IAP) proteins in regulation of inflammation and innate immunity // Discov. Med. — 2011 Mar. — 11(58). — 221-31. — PMID: 21447281.
8. Denko N.C. Investigating hypoxic tumor physiology through gene expression patterns / N.C. Denko, L.A. Fontana, K.M. Hudson, P.D. Sutphin, S. Raychaudhuri, R. Altman, A.J. Giaccia // Oncogene. — 2003, Sep 1. — 22(37). — 5907-14. — doi: 10.1038/sj.onc.1206703.
9. Franek W.R. NF-κB protects lung epithelium against hyperoxia-induced nonapoptotic cell death-oncosis / W.R. Franek, D.M. Morrow, H. Zhu, I. Vancurova, V. Miskolci, K. Darley-Usmar, H.H. Simms, L.L. Mantell // Free Radic. Biol. Med. — 2004, Nov 15. — 37(10). — 1670-9. — doi: 10.1016/j.freeradbiomed.2004.08.007.
10. Haddad J.J. Science review: Redox and oxygen-sensitive transcription factors in the regulation of oxidant-mediated lung injury: role for hypoxia-inducible factor-1α // Crit. Care. — 2003 Feb. — 7(1). — 47-54. — PMCID: PMC154109.
11. Haddad J.J., Harb H.L. Cytokines and the regulation of hypoxia-inducible factor (HIF)-1alpha // Int. Immunopharmacol. — 2005 Mar. — 5(3). — 461-83. — doi: 10.1016/j.intimp.2004.11.009.
12. Heyman S.N., Rosen S., Rosenberger C. Hypoxia-inducible factors and the prevention of acute organ injury // Crit. Care. — 2011. — 15(2). — 209. — doi: 10.1186/cc9991.
13. Janssens S., Tinel A. The PIDDosome, DNA-damage-induced apoptosis and beyond // Cell. Death Differ. — 2012 Jan. — 19(1). — 13-20. — doi: 10.1038/cdd.2011.162.
14. Jiang J., George S.C. TGF-β2 reduces nitric oxide synthase mRNA through a ROCK-dependent pathway in airway epithelial cells // Am. J. Physiol. Lung Cell. Mol. Physiol. — 2011 Sep. — 301(3). — L361-7. — doi: 10.1152/ajplung.00464.2010.
15. Kuwano K. Epithelial cell apoptosis and lung remodeling // Cell. Mol. Immunol. — 2007 Dec. — 4(6). — 419-29. — PMID: 18163953.
16. Kuwano K. Involvement of epithelial cell apoptosis in interstitial lung diseases // Intern. Med. — 2008. — 47(5). — 345-53. — PMID: 18310962.
17. Lago C.U. p53, aerobic metabolism, and cancer / C.U. Lago, H.J. Sung, W. Ma, P.Y. Wang, P.M. Hwang // Antioxid Redox Signal. — 2011, Sep 15. — 15(6). — 1739-48. — doi: 10.1089/ars.2010.3650.
18. Lambeth J.D. NOX enzymes and the biology of reactive oxygen // Nat. Rev. Immunol. — 2004 Mar. — 4(3). — 181-9. — doi:10.1038/nri1312.
19. Lambeth J.D., Neish A.S. Nox enzymes and new thinking on reactive oxygen: a double-edged sword revisited // Ann. Rev. Pathol. — 2014. — 9. — 119-45. — doi: 10.1146/annurev-pathol-012513-104651.
20. Lesur O. Role of interferon-g and interleukin-2 in rat lung epithelial cell migration and apoptosis after oxidant injury // O. Lesur, M. Brisebois, A. Thibodeau, F. Chagnon, D. Lane, T. Füllöp // Am. J. Physiol. Lung. Cell. Mol. Physiol. — 2004 Jan. — 286(1). — L4-L14. — doi: 10.1152/ajplung.00367.2002.
21. Li N., Nel A.E. Role of the Nrf2-mediated signaling pathway as a negative regulator of inflammation: implications for the impact of particulate pollutants on asthma // Antioxid Redox Signal. — 2006 Jan — Feb. — 8(1–2). — 88-98. — doi: 10.1089/ars.2006.8.88.
22. Li N., Xia T., Nel A.E. The Role of Oxidative Stress in Ambient Particulate Matter-induced Lung Diseases and Its Implications in the Toxicity of Engineered Nanoparticles // Free Radic. Biol. Med. — 2008, May 1. — 44(9). — 1689-99. — doi: 10.1016/j.freeradbiomed.2008.01.028.
23. Lian X. Overexpression of Stat 3C in pulmonary epithelium protects against hyperoxic lung injury / X. Lian, Y. Qin, S.A. Hossain, L. Yang, A. White, H. Xu, J.M. Shipley, T. Li, R.M. Senior, H. Du, C. Yan // J. Immunol. — 2005, Jun 1. — 174(11). — 7250-6. — doi: 10.4049/jimmunol.174.11.7250.
24. Majmundar A.J., Wong W.J., Simon M.C. Hypoxia-inducible factors and the response to hypoxic stress // Mol. Cell. — 2010, Oct 22. — 40(2). — 294-309. — doi: 10.1016/j.molcel.2010.09.022.
25. Milla C. Myeloperoxidase deficiency enhances inflammation after allogeneic marrow transplantation / C. Milla, S. Yang, D.N. Cornfield, M.L. Brennan, S.L. Hazen, A. Panoskaltsis-Mortari, B.R. Blazar, I.Y.Haddad // Am. J. Physiol. Lung. Cell. Mol. Physiol. — 2004 Oct. — 287(4). — L706-14.
26. Nath B., Szabo G. Hypoxia and hypoxia inducible factors: diverse roles in liver diseases // Hepatology. — 2012 Feb. — 55(2). — 622-33. — doi: 10.1002/hep.25497.
27. Nordberg J., Arner E.S. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system // Free Radic. Biol. Med. — 2001, Dec 1. — 31(11). — 1287-312. — doi: 10.1016/S0891-5849(01)00724-9.
28. Qin S. Smac: Its role in apoptosis induction and use in lung cancer diagnosis and treatment / S. Qin, C. Yang, S. Li, C. Xu, Y. Zhao, H. Ren // Cancer Lett. — 2012, May 1. — 318(1). — 9-13. — doi: 10.1016/j.canlet.2011.12.024.
29. Reddy S.P. The antioxidant response element and oxidative stress modifiers in airway diseases // Curr. Mol. Med. — 2008 Aug. — 8(5). — 376-83. — PMCID: PMC2828610.
30. Schumacker P.T. Lung cell hypoxia: role of mitochondrial reactive oxygen species signaling in triggering responses // Proc. Am. Thorac. Soc. — 2011 Nov. — 8(6). — 477-84. — doi: 10.1513/pats.201103-032MW.
31. Semenza G.L. Targeting HIF-1 for cancer therapy // Nat. Rev. Cancer. — 2003 Oct. — 3(10). — 721-32. — PMID: 13130303.
32. Shimoda L.A., Semenza G.L. HIF and the lung: role of hypoxia-inducible factors in pulmonary development and disease // Am. J. Respir. Crit. Care Med. — 2011, Jan 15. — 183(2). — 152-6. — doi: 10.1164/rccm.201009-1393PP.
33. Wenger R.H. Cellular adaptation to hypoxia: O2-sensing protein hydroxylases, hypoxia-inducible transcription factors, and O2-regulated gene expression // FASEB J. — 2002 Aug. — 16(10). — 1151-62. — doi: 10.1096/fj.01-0944rev.
34. Xing Z. IL-6 is an antiinflammatory cytokine required for controlling local or systemic acute inflammatory responses / Z. Xing, J. Gauldie, G. Cox, H. Baumann, M. Jordana, X.F. Lei, M.K. Achong // J. Clin. Invest. — 1998, Jan 15. — 101(2). — 311-20. — PMID: 9435302.
35. Zhou J. PI3K/Akt is required for heat shock proteins to protect HIF-1 alpha from pVHL-independent degradation / J. Zhou, T. Schmid, R. Frank, B. Brune // J. Biol. Chem. — 2004, Apr 2. — 279(14). — 13506-13. — doi: 10.1074/jbc.M310164200.

/138-2.jpg)