Журнал «Травма» Том 17, №1, 2016
Посттравматический остеоартрит: воспалительные, клеточные и биомеханические механизмы прогрессирования заболевания
Резюме
Посттравматический остеоартрит (ПТОА) возникает при травме суставов, на его долю приходится значительная часть пациентов с остеоартритом (ОА). Внутрисуставные переломы, сопровождающиеся гемартрозом, разрывы и грубые повреждения менисков, связочного аппарата, травмы хрящевой ткани являются наиболее частыми причинами, ведущими к ПТОА. Повреждение суставного хряща может происходить в период сразу после травмы, в ближайшее время или развиваться через несколько лет после травмы. Даже при соответствующем лечении, таком, как жесткая фиксация внутрисуставных переломов, реконструкция поврежденных связок с успешным восстановлением биомеханики сустава, риск развития ПТОА колеблется от 20 до более чем 50 %. В последнее десятилетие активно обсуждается и изучается роль воспаления в развитии и прогрессировании ПТОА. Признаки воспаления можно наблюдать в суставных жидкостях и тканях у пациентов с травмой суставов и риском развития посттравматического остеоартрита. Кроме того, воспалительные механизмы способствуют риску развития остеоартрита и прогрессированию его после травмы. В развитии ПТОА задействованы молекулярные и клеточные механизмы. Синовиальная оболочка, суставной хрящ и субхондральная кость также реагируют на травму изменением метаболической активности и экспрессии провоспалительных белков. Данный обзор суммирует результаты недавних исследований патологических механизмов, лежащих в основе развития ПТОА. Это позволяет по-новому взглянуть на механизмы развития ПТОА, роль воспаления в прогрессировании болезни, а также оценить потенциальные возможности для раннего фармакологического вмешательства.
Посттравматичний остеоартрит (ПТОА) виникає при травмі суглобів, на його частку припадає значна частина пацієнтів з остеоартритом (ОА). Внутрішньосуглобові переломи, що супроводжуються гемартрозом, розриви і грубі ушкодження менісків, зв’язкового апарату, травми хрящової тканини є найбільш частими причинами, що призводять до ПТОА. Пошкодження суглобового хряща може відбуватися в період відразу після травми, найближчим часом або розвиватися через кілька років після травми. Навіть при відповідному лікуванні, такому, як жорстка фіксація внутрішньосуглобових переломів, реконструкція пошкоджених зв’язок з успішним відновленням біомеханіки суглоба, ризик розвитку ПТОА коливається від 20 до більш ніж 50 %. Останнім десятиліттям активно обговорюється і вивчається роль запалення в розвитку і прогресуванні ПТОА. Ознаки запалення можна спостерігати в суглобових рідинах і тканинах у пацієнтів із травмою суглобів і ризиком розвитку посттравматичного остеоартриту. Крім того, запальні механізми сприяють ризику розвитку остеоартриту і прогресуванню його після травми. У розвитку ПТОА задіяні молекулярні і клітинні механізми. Синовіальна оболонка, суглобовий хрящ і субхондральна кістка також реагують на травму зміною метаболічної активності та експресії прозапальних білків. Даний огляд підсумовує результати недавніх досліджень патологічних механізмів, що лежать в основі розвитку ПТОА. Це дозволяє по-новому поглянути на механізми розвитку ПТОА, роль запалення в прогресуванні хвороби, а також оцінити потенційні можливості для раннього фармакологічного втручання.
Post-traumatic OA (PTOA) arises from joint injury, it accounts for a significant portion of patients with osteoarthritis. Intra-articular fractures associated with hemarthrosis, ruptures and rough damage to the meniscus, ligaments, cartilage injuries are the most common causes of PTOA. Damage to articular cartilage can occur in the period immediately after the injury, in the near future or develop several years after the injury. Even with appropriate treatment, such as rigid fixation of intra-articular fractures, reconstruction of damaged ligament with a successful restoration of joint biomechanics, the risk of PTOA ranges from 20 to more than 50 %. In the last decade, the role of inflammation in the development and progression of PTOA is being actively discussed and studied. Signs of inflammation may be observed in the joint fluid and tissues of patients with injuries of the joints and the risk of post-traumatic osteoarthritis. In addition, inflammatory mechanisms lead to the risk of developing osteoarthritis and its progression after injury. The development of PTOA involves molecular and cellular mechanisms. Synovium, articular cartilage and subchondral bone also react to the injury by changing metabolic activity and expression of inflammatory proteins. This review summarizes the results of recent studies of pathological mechanisms underlying PTOA development. This allows you to take a new look at the mechanisms of PTOA development, the role of inflammation in the progression of the disease, and to assess the limitations for early pharmacological intervention.
Ключевые слова
посттравматический остеоартрит, травма сустава, суставной хрящ, воспаление, цитокины.
посттравматичний остеоартрит, травма суглоба, суглобовий хрящ, запалення, цитокіни.
post-traumatic arthritis, joint injury, articular cartilage, inflammation, cytokines.
Статья опубликована на с. 99-105
Травмы суставов являются установленным фактором развития остеоартрита (ОА) и формируют особый фенотип посттравматического ОА (ПТОА) [2]. Приблизительно 12 % всех случаев ОА связано с предварительной травмой сустава [9]. Внутрисуставные переломы, сопровождающиеся гемартрозом, разрывы и грубые повреждения менисков, связочного аппарата, травмы хрящевой ткани являются наиболее частыми причинами, ведущими к ПТОА. ПТОА отличается от других фенотипов ОА тем, что мы точно знаем время провоцирующего травматического события, что дает возможность понять и оценить события после повреждения сустава, приводящие к прогрессированию ОА. В отличие от возрастзависимого и/или метаболического ОА, развивающегося у людей пожилого и среднего возраста, ПТОА поражает более молодых пациентов и характеризуется довольно быстрой прогрессией [10]. После травмы сустава и хирургических вмешательств по рестабилизации сустава часто возникает нарушение функциональной активности суставов, нестабильность и хронической болевой синдром. Травматическое повреждение сустава с нарушением биомеханики является известным фактором прогрессирующей суставной дегенерации [18], однако оперативные вмешательства по поводу рестабилизации сустава не уменьшают этот риск [14]. Наоборот, в среднем через 20 лет после менискэктомии у 3/4 пациентов развивался тибиофеморальный или пателлофеморальный ОА [37]. После менискэктомии у пациентов в течение длительного времени сохраняется повышенный уровень интерлейкина-6 (ИЛ-6) и фактора некроза опухоли α (ФНО-α) в синовиальной жидкости, что, по-видимому, связано с повышенным риском прогрессирования радиографического ОА [27]. В научной литературе даже появился термин «менискэктомия-индуцированный остеоартрит». Многие исследователи полагают, что молекулярные и клеточные изменения в суставных тканях после травмы являются необратимыми, мало того, запускают каскад биологических реакций, способных в дальнейшем повреждать суставные структуры [29].
Биомеханические факторы всегда рассматривались как ведущие в этиопатогенезе ОА: патологические изменения в суставном хряще возникали либо из-за анормальной (чрезмерной) нагрузки, действующей в условиях нормальной физиологии, либо из-за нормальной нагрузки, действующей в присутствии аномальной физиологии [21]. На рис. 1 представлено влияние механической нагрузки на развитие остеоартрита. Механически-индуцированное повреждение суставных структур и травмы сустава приводят к персистирующим воспалительным реакциям, усиливая структурные повреждения суставного хряща [18] (рис. 2).
/100.jpg)
Согласно парадигме 80-х годов прошлого столетия, остеоартроз рассматривали как болезнь изнашивания тканей (wear and tear), то есть как дегенеративное заболевание, при котором происходит замедление репаративных процессов в поврежденном хряще в результате биомеханических и биохимических изменений в суставе [7]. Эта парадигма базировалась на наблюдениях и экспериментальных данных того времени, согласно которым хондроциты имеют низкую метаболическую активность и не способны восстановить поврежденный хрящ. Вследствие отсутствия васкуляризации хрящ также не может ответить на раздражающие или повреждающие стимулы обычной воспалительной реакцией. Поэтому ранее заболевание расценивалось как стигма старения организма в целом и сопутствующей этому процессу дегенерации суставных структур в частности. Эти воззрения легли в основу одного из определений остеоартроза: остеоартроз относится к группе первично невоспалительных заболеваний суставов различной этиологии и может рассматриваться как анатомо-клинический синдром, который характеризуется болью механического типа у лиц старше 45 лет с соответствующими рентгенологическими данными [4]. Поэтому термин «остеоартроз» соответствовал старым представлениям об этом заболевании.
В последние годы четко доказано, что такой взгляд является ошибочным: ОА не просто болезнь износа (болезнь wear and tear), а скорее анормальное ремоделирование суставных тканей, управляемое множеством провоспалительных факторов, продуцируемых прежде всего субхондральной костью и синовией [5]. По мере изучения патогенеза заболевания, внедрения новых методов диагностики стало ясно, что ОА характеризуется хроническим воспалением, при котором в патологический процесс вовлечены все компоненты сустава, включая синовиальную оболочку, хрящ, суставную капсулу, связки, сухожилия, субхондральную кость [1].
Спор о возможном только прямом механическом повреждении суставного хряща был фактически прекращен после открытия внутриклеточного механосигналинга [21]. Оказалось, что любое ненормальное механическое напряжение — растяжение, сжатие, компрессия, напряжение сдвига, гидростатическое давление — может быть преобразовано во внутриклеточные сигналы посредством возбуждения механорецепторов, расположенных на поверхности клеток сустава. Эти сигналы могут привести к избыточной экспрессии провоспалительных медиаторов, таких как простагландины, цитокины, хемокины, в хондроцитах и клетках субхондральной кости, а также изменить сам фенотип хондроцитов и остеобластов [39]. Преобразование механического сигналинга в синтез медиаторов воспаления опосредуется активацией внутриклеточных сигнальных путей, прежде всего через NF-kВ и МАРК [6].
В последнее десятилетие проблема ПТОА активно изучается, особенно острая посттравматическая фаза, с использованием разнообразных моделей на животных, животных и человеческих культур тканей. Более глубокое понимание механобиологии, молекулярных и клеточных процессов, приводящих к деградации хряща в относительно ранние фазы после разрешения травматического повреждения, потенциально открывает новые возможности для раннего клинического вмешательства.
Наиболее общими травмами, приводящими к развитию ПТОА коленного сустава, является разрыв передней крестообразной связки (ПКС), повреждение (полный и/или частичный разрыв) мениска и внутрисуставной перелом. Эти травматические повреждения приводят к гемартрозу, повреждению и апоптозу хондроцитов и остеобластов, а также высвобождению большого количества медиаторов воспаления (рис. 2).
Патологические изменения в синовии
Недавние исследования синовиальной жидкости у 111 пациентов (средний возраст 27 лет) с травматическим повреждением ПКС продемонстрировали высокие уровни ИЛ-1β, -6, -8 и ФНО-a в первый день после травмы, однако показатели этих цитокинов долгое время (до 25-го дня) оставались повышенными в сравнении с пациентами без травмы [40]. Концентрации маркеров разрушения протеогликанов (остеопонтин, сульфатированные гликозаминогликаны, SPARC) отставали от провоспалительных цитокинов: их показатели значительно возрастали на второй день после травмы. При сопутствующем внутрисуставном переломе наиболее весомо возрастали концентрации ИЛ-8 и ФНО-a.
Концентрация противовоспалительных цитокинов в синовиальной жидкости также возрастает после травмы [35]. В первые 2 недели после повреждения ПКС коленного сустава регистрировались более высокие показатели ИЛ-10, однако впоследствии его концентрация снижалась [24, 35]. В экспериментальных исследованиях именно ИЛ-10 совместно с ИЛ-4 защищает суставной хрящ от последствий активации воспаления в ответ на кровоизлияние в сустав, что свидетельствует о возможной хондропротекторной роли этих цитокинов при гемартрозе [42]. Уровни ИЛ-1Ra также возрастают в синовиальной жидкости после острой травмы ПКС, но в течение следующих 3–6 недель снижаются до уровня нормы. Однако именно ИЛ-1Ra может блокировать негативные эффекты ИЛ-1 в поврежденном суставе [13].
После острой фазы концентрация провоспалительных цитокинов постепенно снижается, хотя некоторые авторы сообщили о сохранении их повышенного уровня от нескольких месяцев до нескольких лет после травмы сустава. Так, через 6 месяцев после разрыва ПКС коленного сустава отмечалось удержание высоких концентраций ИЛ-1β в синовиальной жидкости по сравнению с контрольной группой, а уровень повышения коррелировал (r2 = 0,954) со степенью хрящевых повреждений [30]. Другие исследователи сообщают, что в синовиальной жидкости уровень ИЛ-1 снижается в первую очередь, а концентрации ИЛ-6 и ФНО-a остаются повышенными в течение длительного времени (более 6 месяцев после травмы) [17]. Одновременно после травмы происходит снижение концентрации лубрицина (протеогликана 4) в синовиальной жидкости, что может обусловить возрастание риска повышенного износа сустава вследствие уменьшения вязкоупругих свойств синовиальной жидкости. Лубрицин вырабатывается хондроцитами и клетками синовиальной оболочки, присутствует в синовиальной жидкости и на поверхности (поверхностный слой) суставного хряща и, следовательно, играет важную роль в поддержании стабильности суставной смазки и синовиального гомеостаза [3]. После травмы его низкий уровень удерживается до 12 мес. [44]. Снижение концентрации лубрицина коррелирует с уровнем ФНО, а ингибирование ФНО приводит к повышению концентрации лубрицина [16, 17]. Концентрация других суставных лубрикантов, таких как гиалуроновая кислота, общие протеогликаны, олигомерный матриксный протеин хряща, также снижается после травмы, а восстановление нормальных уровней тормозится высокими концентрациями ФНО, ИЛ-1β и тромбоцитарного фактора роста β (TGF-β) [8].
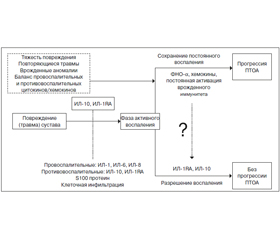
В подострой и хронической фазе (от 2 месяцев до 1 года) посттравматического периода концентрация провоспалительных цитокинов (ИЛ-6, -8, ФНО-a) остается существенно повышенной по сравнению с контролем. Гипотезируется, что в остром периоде после травмы формируется дисбаланс про- и противовоспалительных цитокинов, вследствие чего провоспалительный ответ у некоторых пациентов недостаточно контролируется противовоспалительной системой, что обусловливает хронизацию воспаления и приводит к ПТОА [29] (рис. 3).
/102.jpg)
Патологические процессы после повреждений суставов могут колебаться в зависимости от степени механического воздействия на ткани сустава. Низкоэнергетические травмы, такие как ушибы, дислокации, повреждения связок и менисков, обычно вызывают повреждение суставной поверхности без перелома костей, однако могут возникать микротрещины хряща и субхондральной кости. Высокоэнергетические травмы ассоциируются с внутрисуставными переломами и более грубыми повреждениями суставного хряща и субхондральной кости. В острой посттравматической фазе эффекты травмы включают структурное повреждение тканей сустава, гемартроз и апоптоз хондроцитов/остеобластов [41]. В экспериментальных работах продемонстрировано, что сила удара 1,43 Дж (высокоэнергетическая травма) вызывает гибель всех хондроцитов. При силе удара 0,71 Дж выживают 73 % хондроцитов, при силе удара 1,07 Дж — 64 %. Эти данные указывают, что ранняя хондропротекторная терапия может замедлить процесс апоптоза хондроцитов. Также в момент травмы синовиальная жидкость разбавляется вследствие внутрисуставного кровотечения/кровоизлияния в сустав, экстравазации плазмы, что существенно снижает концентрацию гиалуроновой кислоты и вискозо-эластичных свойств синовиальной жидкости. Травма сустава вызывает подавление синтеза коллагена и протеогликанов в суставном хряще. Оставшиеся жизнеспособные клетки отвечают на травму повышенной синтетической активностью и сверхэкспрессией матричных металлопротеиназ (ММП) и провоспалительных медиаторов [28].
В хронической фазе метаболические изменения в суставном хряще и других суставных тканях медленно прогрессируют, сменяя клинически бессимптомный период симптоматической фазой с болью в суставах и дисфункцией вследствие деструктивных изменений. Длительное время ПТОА является бессимптомным заболеванием, тогда же, когда возникают выраженные клинические симптомы, пациентам остается только хирургическое лечение: артропластика, остеотомия, артродез, тотальное эндопротезирование. Однако при разработке эффективных терапевтических стратегий на ранних этапах после травмы возможно избежать проведения хирургических вмешательств. Для этого необходимо и важно понять, какие метаболические изменения на клеточном и молекулярном уровнях происходят в острой посттравматической фазе, а также в период бессимптомного протекания ПТОА.
На роли воспаления в развитии ПТОА акцентируется во многих клинических и экспериментальных исследованиях. Ниже суммированы основные особенности воспалительной реакции после травмы, что подчеркивает роль воспаления в формировании ПТОА [29].
– Признаки воспаления выявляются в ранние сроки после повреждения сустава.
– Характерно низкоуровневое воспаление.
– Модели воспаления могут меняться со временем после травмы.
– Степень/тяжесть первоначальной травмы влияет на выраженность синовиального воспаления.
– Все ткани сустава вовлечены в воспаление после травмы, поэтому различные типы клеток могут поддерживать воспалительный процесс в суставе.
– Воспаление может произойти даже в отсутствие нестабильности сустава после травмы.
– Воспаление способствует дальнейшему повреждению хряща.
– Воспаление является модифицируемой особенностью ПTOA.
Механобиологические механизмы
Механическое повреждение и разрушение внеклеточного матрикса приводит к высвобождению гликозаминогликанов, накоплению обрывков коллагена, которые воздействуют на хондроциты через механорецепторы и молекулярные рецепторы, приводя к изменению экспрессии генов, а в дальнейшем к деградации суставного хряща [20]. Механическое воздействие активирует α5β1 интегрин-зависимый внутриклеточный сигнальный каскад, конечным эффектом чего является высвобождение ИЛ-4. Механическая компрессия активирует сигнальные пути с участием высвобождения внутриклеточного кальция, а также активацию циклической АМФ-активированной протеинкиназы А. В остром посттравматическом периоде также активируются известные транскрипционные факторы C-fos и c-jun [19]. Несмотря на то, что многие механизмы предстоит еще выяснить, очевидно, что анормальная механическая стимуляция может активировать механорецепторы хондроцитов и катаболические пути, приводящие к деградации экстрацеллюлярного матрикса [25].
Клеточные механизмы
Многочисленные исследования in vitro и in vivo позволили идентифицировать смерть хондроцитов после механического воздействия на суставной хрящ [15, 32]. Это происходит за счет как некроза клеток, так и апоптоза. Гибель клеток вследствие некроза может происходить в момент механического воздействия при силе соударения больше чем 15–20 МПа (непосредственно под воздействующей силой). Количество некротизированных хондроцитов предсказуемо увеличивается при возрастании силы удара до 35 МПа. При воздействии с силой более 40 МПа наблюдается полная гибель клеток [32]. B. Kurz и соавт. (2001) [26] обнаружили, что скорость деформации также является важным фактором, определяющим ответ хондроцита на повреждение. Более высокая скорость деформации ассоциируется со снижением анаболических процессов и жизнеспособности клеток. Поскольку хондроциты являются основными клетками, ответственными за поддержание функций суставного хряща, то их смерть рассматривается как центральный элемент в развитии ПТОА [12, 31]. Однако апоптоз хондроцитов может происходить и в более поздние временные промежутки после суставной травмы [43]. Апоптоз хондроцитов коррелирует с повреждением экстрацеллюлярного матрикса, что возникает на более поздних фазах после травмы вследствие активации патологических сигнальных путей, изменения экспрессии генов, накопления в синовиальной жидкости провоспалительных цитокинов, активации матриксных протеиназ. Кроме того, некоторые экспериментальные работы указывают на апоптоз хондроцитов под влиянием свободных радикалов, активно высвобождающихся из митохондрий [31]. Снижения апоптоза можно достичь применением антиоксидантов после травмы сустава. Однако данный механизм вряд ли будет первопричиной изменений при разрыве связок или повреждении менисков; скорее всего, именно гибель хондроцитов как инициальный механизм играет ведущую роль при внутрисуставных переломах и ударах значительной силы.
Молекулярные и метаболические механизмы
В остром периоде после травмы происходит существенное изменение экспрессии матриксных ферментов, разрушающих суставной хрящ. В течение первых 4 часов после травмы резко возрастает экспрессия матриксной металлопротеиназы 3 (ММП-3, стромелизин 1), дезинтегрина, металлопротеиназы с тромбоспондином 5 (ADAMTS-5) и тканевого ингибитора металлопротеиназы [28]. Все эти ферменты приводят к разрушению эктрацеллюлярного матрикса хряща. При этом ADAMTS-5 вызывает наименьшие изменения в субхондральной кости и суставном хряще в сравнении с другими матричными энзимами. В исследовании I. Polur и соавт. (2010) [38] показано, что после дестабилизации медиального мениска значительно повышается экспрессия белка HTRA1, который, как известно, регулирует доступность и активность инсулиноподобных факторов роста. Этот белок также вовлечен в процессы деградации экстрацеллюлярного матрикса. После травмы также происходит высвобождение коллагена 2-го типа и разрушение протеогликанов [34]. Коллагеновые молекулы затем воздействуют на рецепторный домен (Ddr2) через ras/raf/MEK/ERK и сигнальные пути р38, индуцируя повышенную экспрессию ММП-13.
Несколько исследований подтверждают факт индукции коллагеном 2-го типа ММП-1, -2, -13, -14, а также –ИЛ-1β, -6 и -8, а также активацию сигнальных путей митоген-активированной протеинкиназы р38 (МАРК р38) и ядерного фактора каппа В (NFkB). Таким образом, формируется порочный круг, в котором ММП разрушают экстрацеллюлярный матрикс, повреждая хондроциты и высвобождая большое количество коллагена 2-го типа, что опять же стимулирует выработку матриксных ферментов, разрушающих хрящ.
Исследования ранней посттравматической фазы показали повышенную транскрипцию молекул, участвующих как в катаболических, так и в анаболических процессах. Суставной хрящ при ПТОА характеризуется протеогликановой недостаточностью, повышенным содержанием воды и снижением жесткости экстрацеллюлярного матрикса [11, 36]. Существуют доказательства повышения анаболических процессов, что предполагает репаративные реакции на повреждение хряща.
Известно, что суставной хрящ взрослых людей является бессосудистой тканью. Это важно для функции хряща, окружения хондроцитов и функционирования экстрацеллюлярного матрикса. Фактор роста эндотелия сосудов (VEGF) индуцируется повреждением суставного хряща [23]. Повышенная экспрессия VEGF сопровождается пониженной экспрессией хондромодулина 1, антиангиогенного фактора, участвующих в поддержании функционирования лишенного сосудов суставного хряща [22].
Таким образом, механическая травма вызывает дисрегуляцию многочисленных сигнальных путей, изменяет экспрессию генов, нарушает баланс между катаболическими и анаболическими процессами на молекулярном уровне.
Патологические изменения кости
Как известно, прогрессирование ОА ассоциируется с поражением субхондральной кости и развитием краевых остеофитов [2]. Серьезные травмы сустава, прежде всего внутрисуставные переломы, очевидно, повреждают субхондральную кость, а разрывы связок ведут к костным ушибам и микротрещинам [33]. Экспериментальные исследования подтвердили гистологические и микроархитектурные изменения субхондральной кости после травмы. Эти данные указывают на существование тесной взаимосвязи субхондральной кости и суставного хряща, однако изменения в кости происходят значительно позже после травмы.
Активация воспалительных механизмов имеет решающее значение для развития ПТОА и является частью патологических реакций, какими сустав реагирует на травму. В посттравматическом периоде наблюдается повышение продукции провоспалительных медиаторов (цитокинов и хемокинов), клеточная инфильтрация, активация сигнальных путей, что является различными аспектами общей воспалительной реакции. Эти аспекты воспроизведены в многочисленных экспериментальных моделях на животных и подтверждены клинически. Эти модели подтвердили факт раннего развития воспалительной реакции, которая со временем изменяется и модифицируется (рис. 3).
Таким образом, ПТОА этиологически связан с травмой сустава и развивается после нее. Даже при оказании хирургической помощи риск ПТОА колеблется от 20 до более чем 50 % [25]. Часовые интервалы развития ПТОА сильно варьируют, однако патологические изменения в синовии, суставном хряще и субхондральной кости происходят в первые часы после травмы и могут удерживаться до 12 месяцев. В развитие и прогрессирование ПТОА, несомненно, вовлечены и традиционные факторы прогрессирования ОА: возраст, ожирение, гормональные сдвиги и другое, утяжеляя клиническую симптоматику ОА, влияя на выраженность воспалительной и клеточной реакции. Данный обзор суммирует результаты недавних исследований патологических механизмов, лежащих в основе развития ПТОА. Это позволяет по-новому взглянуть на механизмы развития ПТОА, роль воспаления в прогрессировании болезни, а также оценить потенциальные возможности для раннего фармакологического вмешательства.
Список литературы
Список литературы находится в редакции
1. Golovach I.Yu. Osteoarthrit: sovremennye fundamentalnie i prikladnye asperity pathogeneza zabolevaniya. Bol. Sustavy. Pozvonochnik. 2014; 3 (15): 54-58.
2. Kovalenko V.N., Bortkevych O.P. Osteoarthroz. Praktychna nastanova. – 3 vydannya zi zminamy. – K.: Morion, 2010. - 607 s.
3. Sinyachenko O.V. Sovremennye fspekty analiza sinovialnoj zhidkosti. Ukr. Revmatol. Zhurnal. 2008; 2 (32): 30-39.
4. Khitrov N.A. Osteoarthros I osteoarthrit – ot novykh vzglyadov na patogenez k novomu nazvaniyu. Meditsinskiy sovet. 2013; 4: 74-78.
5. Berenbaum F. Osteoarthritis as an inflammatory disease (osteoarthritis is not osteoarthrosis!). Osteoarthritis and Cartilage. 2013, 21: 16-21. doi: 10.1016/j.joca.2012.11.012.
6. Berenbaum F. Signaling transduction: target in osteoarthritis. Curr. Opin. Rheumatol. 2004; 16(5): 616-622.
7. Bijlsma J.W., Berenbaum F., Lafeber F.P. Osteoarthritis: an update with relevance for clinical practice. Lancet. 2011; 377: 2115-2126. doi: 10.1016/S0140-6736(11)60243-2.
8. Blewis M.E., Lao B.J., Schumacher B.L., Bugbee W.D., Sah R.L., Firestein G.S. Interactive cytokine regulation of synoviocyte lubricant secretion. Tissue Eng Part A 2010; 16: 1329-1337. doi: 10.1089/ten.TEA.2009.0210.
9. Brown T.D., Johnston R.C., Saltzman C.L., Marsh J.L., Buckwalter J.A. Posttraumatic osteoarthritis: a first estimate of incidence, prevalence, and burden of disease. J. Orthop. Trauma. 2006; 20: 739-744.
10. Buckwalter J.A. Articular cartilage: injuries and potential for healing. J. Orthop. Sports. Phys. Ther. 1998; 28: 192-202. doi: 10.2519/jospt.1998.28.4.192
11. Buckwalter J.A., Brown T.D. Joint injury, repair, and remodeling: roles in post-traumatic osteoarthritis. Clin. Orthop. Relat. Res. 2004; 423: 7-16.
12. Buckwalter J.A., Mankin H.J., Grodzinsky A.J. Articular cartilage and osteoarthritis. Instr. Course Lect. 2005; 54: 465-480.
13. Cameron M.L., Fu F.H., Paessler H.H., Schneider M., Evans C.H. Synovial fluid cytokine concentrations as possible prognostic indicators in the ACL-deficient knee. Knee Surg. Sports Traumatol. Arthrosc.1994; 2: 38-44.
14. Chalmers P.N., Mall N.A., Moric M., et al. Does ACL reconstruction alter natural history?: a systematic literature review of long-term outcomes. J Bone Joint Surg Am 2014; 96: 292-300. doi: 10.2106/JBJS.L.01713.
15. D'Lima D.D., Hashimoto S., Chen P.C., Colwell C.W.Jr., Lotz M.K. Impact of mechanical trauma on matrix and cells. Clin. Orthop. Relat. Res. 2001; 391 Suppl.: S90-99.
16. Elsaid K.A., Fleming B.C., Oksendahl H.L., et al. Decreased lubricin concentrations and markers of joint inflammation in the synovial fluid of patients with anterior cruciate ligament injury. Arthritis Rheum. 2008; 58: 1707-1715. doi: 10.1002/art.23495.
17. Elsaid K.A., Machan J.T., Waller K., Fleming B.C., Jay G.D. The impact of anterior cruciate ligament injury on lubricin metabolism and the effect of inhibiting tumor necrosis factor alpha on chondroprotection in an animal model. Arthritis Rheum 2009; 60: 2997-3006. doi: 10.1002/art.24800.
18. Felson D.T. Osteoarthritis as a disease of mechanics. Osteoarthritis Cartilage. 2013; 21: 10-15. doi: 10.1016/j.joca.2012.09.012.
19. Fitzgerald J.B., Jin M., Dean D., Wood D.J., Zheng M.H., Grodzinsky A.J. Mechanical compression of cartilage explants induces multiple time-dependent gene expression patterns and involves intracellular calcium and cyclic AMP. J Biol Chem 2004; 279: 19502-19511. doi: 10.1074/jbc.M400437200
20. Grodzinsky A.J., Levenston M.E., Jin M., Frank E.H. Cartilage tissue remodeling in response to mechanical forces. Ann. Rev. Biomed. Eng. 2000; 2: 691-713. doi: 10.1146/annurev.bioeng.2.1.691
21. Guilak F. Biomechanical factors in osteoarthritis. Best Pract. Res. Clin. Rheumatol. 2011; 25(6): 815-823. doi: 10.1016/j.berh.2011.11.013.
22. Hayami T., Funaki H., Yaoeda K., et al. Expression of the cartilage derived antiangiogenic factor chondromodulin-I decreases in the early stage of experimental osteoarthritis. J. Rheumatol. 2003; 30: 2207-2217.
23. Inoue K., Masuko Hongo K., Okamoto M., Nishioka K. Induction of vascular endothelial growth factor and matrix metalloproteinase-3 (stromelysin) by interleukin-1 in human articular chondrocytes and synoviocytes. Rheumatol. Int. 2005; 26: 93-98.
24. Irie K., Uchiyama E., Iwaso H. Intraarticular inflammatory cytokines in acute anterior cruciate ligament injured knee. Knee. 2003; 10: 93-96. doi: http://dx.doi.org/10.1016/S0968-0160(02)00083-2.
25. Kramer W.C., Hendricks K.J., Wang J. Pathogenetic mechanisms of posttraumatic osteoarthritis: opportunities for early intervention. Int. J. Clin. Exp. Med. 2011; 4(4): 285-298.
26. Kurz B., Jin M., Patwari P., Cheng D.M., Lark M.W., Grodzinsky A.J. Biosynthetic response and mechanical properties of articular cartilage after injurious compression. J. Orthop. Res. 2001; 19: 1140-1146. doi: 10.1016/S0736-0266(01)00033-X
27. Larsson S., Englund M., Struglics A., Lohmander L.S. Interleukin-6 and tumor necrosis factor alpha in synovial fluid are associated with progression of radiographic knee osteoarthritis in subjects with previous meniscectomy. Osteoarthritis Cartilage. 2015; 23(11): 1906-1914. doi: 10.1016/j.joca.2015.05.035.
28. Lee J.H., Fitzgerald J.B., Dimicco M.A., Grodzinsky A.J. Mechanical injury of cartilage explants causes specific time-dependent changes in chondrocyte gene expression. Arthritis Rheum 2005; 52: 2386-2395. doi: 10.1002/art.21215
29. Lieberthal J., Sambamurthy N., Scanzello C.R. Inflammation in joint injury and post-traumatic osteoarthritis. Osteoarthritis Cartilage. 2015; 23: 1825-1834. doi: 10.1016/j.joca.2015.08.015.
30. Marks P.H., Donaldson M.L. Inflammatory cytokine profiles associated with chondral damage in the anterior cruciate ligament-deficient knee. Arthroscopy. 2005; 21: 1342-1347. doi: http://dx.doi.org/10.1016/j.arthro.2005.08.034
31. Martin J.A., Buckwalter J.A. Post-traumatic osteoarthritis: the role of stress induced chondrocyte damage. Biorheology. 2006; 43(3-4): 517-521.
32. Milentijevic D., Rubel I.F., Liew A.S., Helfet D.L., Torzilli P.A. An in vivo rabbit model for cartilage trauma: a preliminary study of the influence of impact stress magnitude on chondrocyte death and matrix damage. J. Orthop. Trauma. 2005; 19: 466-473.
33. Mrosek E.H., Lahm A., Erggelet C., Uhl M., Kurz H., Eissner B., Schagemann J.C. Subchondral bone trauma causes cartilage matrix degeneration: an immunohistochemical analysis in a canine model. Osteoarthritis Cartilage 2006; 14: 171-178. doi: http://dx.doi.org/10.1016/j.joca.2005.08.004
34. Nielsen R.H., Stoop R., Leeming D.J., Stolina M., Qvist P., Christiansen C., Karsdal M.A. Evaluation of cartilage damage by measuring collagen degradation products in joint extracts in a traumatic model of osteoarthritis. Biomarkers. 2008; 13: 79-87. DOI:10.1080/13547500701615108
35. Olson S.A., Horne P., Furman B., Huebner J., Al-Rashid M, Kraus V.B., Guilak F. The role of cytokines in posttraumatic arthritis. J. Am. Acad. Orthop. Surg. 2014; 22(1): 29-37. doi: 10.5435/JAAOS-22-01-29.
36. Otsuki S., Brinson D.C., Creighton L., Kinoshita M., Sah R.L., D'Lima D., Lotz M. The effect of glycosaminoglycan loss on chondrocyte viability: a study on porcine cartilage explants. Arthritis Rheum 2008; 58: 1076-1085. doi: 10.1002/art.23381.
37. Paradowski P.T., Lohmander L.S., Englund M. Osteoarthritis of the knee after meniscal resection: long term radiographic evaluation of disease progression. Osteoarthritis Cartilage. 2015; 24: 1424-1417. doi: 10.1016/j.joca.2015.12.002.
38. Polur I., Lee P.L., Servais J.M., Xu L., Li Y. Role of HTRA1, a serine protease, in the progression of articular cartilage degeneration. Histol. Histopathol. 2010; 25: 599-608.
39. Sanchez C., Pesesse L., Gabay O., et al. Regulation of subchondral bone osteoblast metabolism by cyclic compression. Arthritis Rheum. 2012; 64(4): 1193-1203. doi: 10.1002/art.33445.
40. Sward P., Frobell R., Englund M., Roos H., Struglics A. Cartilage and bone markers and inflammatory cytokines are increased in synovial fluid in the acute phase of knee injury (hemarthrosis) - a cross-sectional analysis. Osteoarthritis Cartilage 2012; 20: 1302-1308. doi: 10.1016/j.joca.2012.07.021.
41. Szczodry M., Coyle C.H., Kramer S.J., Smolinski P., Chu C.R. Progressive chondrocyte death after impact injury indicates a need for chondroprotective therapy. Am. J. Sports. Med. 2009; 37: 2318-2322. doi: 10.1177/0363546509348840.
42. van Meegeren M.E., Roosendaal G., Jansen N.W., et al. IL-4 alone and in сombination with IL-10 protects against blood-induced cartilage damage. Osteoarthritis Cartilage.2012; 20:.764-772. doi: 10.1016/j.joca.2012.04.002.
43. Vrahas M.S., Mithoefer K., Joseph D. The long-term effects of articular impaction. Clin. Orthop. Relat. Res. 2004; 423: 40-43.
44. Wei L., Fleming B.C., Sun X., et al. Comparison of differential biomarkers of osteoarthritis with and without posttraumatic injury in the Hartley guinea pig model. J. Orthop. Res. 2010; 28: 900-906. doi: 10.1002/jor.21093.