Статья опубликована на с. 124-127
Синдром впервые описан американским педиатром-эндокринологом Di George Angelo Mario в 1965 году. Синдром Ди Джорджи (СДД) (МКБ-Х — D82.1) является врожденным заболеванием иммунной системы. Клинически данный синдром определяется как первичный иммунодефицит с дефектом Т-клеточного звена иммунитета в комбинации с гипо- или аплазией паращитовидных желез, врожденными пороками сердца и частым выявлением мальформаций лицевой части черепа. У большинства пациентов наблюдаются задержка физического и психомоторного развития, нарушение когнитивных функций.
Заболевание развивается в результате нарушения эмбриогенеза 3–4 глоточных карманов, из которых в норме на 6-й неделе внутриутробного развития формируются тимус и паращитовидные железы. Поражаются и соседние органы, что в дальнейшем выражается дисморфизмом лица: гипоплазией нижней челюсти, короткой верхней губой, низким расположением и деформацией ушных раковин и др.
У 2/3 детей с СДД отмечаются комбинированные врожденные пороки сердца (ВПС) — общий артериальный ствол, тетрада Фалло, удвоение дуги аорты, коарктация аорты, дефект межжелудочковой перегородки. Частыми также являются аномалии крупных сосудов. Формирование данных пороков можно отметить еще до рождения ребенка при ультразвуковом исследовании плода [1, 2].
Впервые в 1981 году было высказано предположение о роли делеции сегмента q-плеча 22-й хромосомы в возникновении синдрома Ди Джорджи. С началом использования метода флуоресцентной гибридизации (FISH in situ) эта хромосомная аномалия идентифицируется более чем у 90 % больных с синдромом Ди Джорджи. Литературные данные последних десяти-пятнадцати лет свидетельствуют о многообразии дислокации генетических изменений при подобной комбинации клинических проявлений. Описаны случаи симптомокомплекса синдрома Ди Джорджи при локализации делеции других хромосом — 10р13, 17р13, 18q21. При наличии классической делеции 22q11.2 используют термин «синдром делеции 22q11.2-хромосомы» или «синдром Ди Джорджи» [3–5].
Возможными факторами риска появления делеции являются сахарный диабет у матери, воздействие алкоголя и вирусных инфекций в первом триместре беременности. Есть данные, что поврежденная 22-я хромосома может наследоваться по аутосомно-доминантному типу, то есть заболевание передается от одного из родителей.
Синдром микроделеции 22q11.2 сочетает гетерогенную группу заболеваний, имеющих в основе одинаковую хромосомную перестройку. Перестройка хромосомы 22 является наиболее частой хромосомной аномалией у детей с ВПС. По данным разных авторов, до 5 % детей с ВПС имеют такую делецию. Частота данной патологии составляет 1 : 4000 новорожденных [2, 3].
Раннее выявление микроделеции 22q11.2 у детей с ВПС очень важно, так как такие пациенты имеют повышенный риск развития гипокальциемии, иммунодефицита, послеоперационных осложнений, асфиксии, синдрома внезапной смерти, трудностей вскармливания, а в старшем возрасте — речевых нарушений, трудностей в обучении и умственной отсталости. Выявление микроделеции 22q11.2 у пациентов с ВПС позволяет скорректировать тактику пред- и послеоперационного ведения таких больных, а в дальнейшем проводить профилактику и коррекцию других патологических проявлений, свойственных микроделеции 22q11.2. Для медико-генетического консультирования важно, что около 10 % случаев микроделеции 22q11.2 являются семейными [6].
СДД представляет собой недостаточность преимущественно клеточного иммунитета. В структуре всех первичных иммунодефицитов его доля составляет 20–25 %. Данный синдром обусловлен первичным нарушением пролиферации и дифференцировки B- и T-лимфоцитов. Однако «полный» синдром Ди Джорджи с выраженными аномалиями иммунной системы встречается крайне редко.
Диагноз ставится на основе клинической картины, а также данных лабораторных показателей. При аплазии или гипоплазии вилочковой железы появляются дефектные Т-лимфоциты, в реакции бласттрансформации они не дают ответа на фитогемагглютинин, отсутствуют Т-супрессоры. Показатели гуморального иммунитета у больных с СДД чаще сохранены. У некоторых больных повышена концентрация IgE, что, возможно, связано с отсутствием Т-супрессоров [1]. Дети с СДД подвержены различным инфекциям, прежде всего дыхательных и мочевыводящих путей. Выраженность иммунодефицита определяет частоту и тяжесть рекуррентных заболеваний. Считают, что с возрастом функция Т-лимфоцитов восстанавливается и к 5 годам проявления иммунологических нарушений могут быть купированы [1, 4].
Клинически наиболее постоянным проявлением заболевания является гипопаратиреоз, в связи с чем наиболее частым дебютом СДД являются тонико-клонические судороги в периоде новорожденности вследствие гипокальциемии. Длительно сниженный уровень кальция в крови может обусловливать нарушения роста и развития ребенка, патологию костной системы, множественный кариес зубов. В более позднем периоде жизни гипокальциемия может стать причиной вторичного синдрома удлиненного интервала QT, желудочковых аритмий и внезапной сердечной смерти [7, 8]. Симптоматическая гипокальциемия в значительной мере осложняет течение ВПС.
Таким образом, на прогноз течения СДД оказывает влияние уровень выраженности дефектов сердечно-сосудистой и эндокринной систем, при «полном» синдроме — степень иммунодефицита. Ввиду многообразия клинических симптомов пациенты с СДД могут наблюдаться врачами разных специальностей.
Собственное наблюдение. Под нашим наблюдением в течение года находился ребенок Ш. 2013 года рождения с диагнозом «синдром Ди Джорджи». Пациент неоднократно поступал в кардиопульмонологическое отделение ДГКБ № 2 г. Днепропетровска на плановое обследование в связи с множественными врожденными пороками развития: ВПС (атрезия легочной артерии 2-го типа с выраженной гипоплазией ствола и веток легочной артерии, подаортальный дефект межжелудочковой перегородки (ДМЖП). Большие аортолегочные коллатеральные артерии. Недостаточность кровообращения 1-й ст.). Врожденный порок развития легких — кистозная гипоплазия левого легкого. Трахеальный бронх справа. Субсегментарный ателектаз верхней доли справа.
Из анамнеза жизни известно, что ребенок родился от 2-й беременности, протекавшей на фоне нефропатии, первых родов в сроке 40 недель. При рождении показатели роста и массы тела ребенка выше среднего (4100 г, рост 55 см). Оценка по шкале Апгар — 8/9 баллов. Ранний неонатальный период протекал с синдромом повышенной нервно-рефлекторной возбудимости, частыми срыгиваниями, гемодинамическими расстройствами. Наблюдалось снижение сатурации кислорода в покое до 88–90 %, при физической нагрузке — до 68 %.
В ходе обследования были диагностированы врожденный порок сердца и сосудов (ДМЖП, атрезия легочной артерии) и субсегментарный ателектаз верхней доли правого легкого. Тогда же были выявлены гипокальциемия, калий-натриевый дисбаланс. Отмечалась выраженная лимфопения при повторных гематологических исследованиях (в пределах 22–24 %). Был заподозрен синдром Ди Джорджи. Исследование паратиреоидного гормона проводилось однократно и не выявило отклонений от нормативных значений (паратгормон — 11,79 пг/мл).
Однако с учетом доминирования в клинической картине симптоматики ВПС ребенок был направлен на консультацию в научно-практичный медицинский центр детской кардиологии и кардиохирургии (НПМЦ ДКК) МЗ Украины. В НПМЦ ДКК в результате проведенных эхокардиографического и томографического исследований был подтвержден диагноз ВПС: атрезия легочной артерии 2-го типа с выраженной гипоплазией ствола и веток легочной артерии; подаортальный ДМЖП; большие аорто-легочные коллатеральные артерии. На ЭхоКГ была отмечена умеренная декстрапозиция аорты. С учетом сложной анатомии врожденного порока его хирургическая коррекция была перенесена на 3 месяца.
При проведении компьютерной томографии были также выявлены морфологические изменения в легких: субсегментарный ателектаз верхней доли справа, трахеальный бронх справа и неравномерность пневматизации легочной паренхимы в нижней доле левого легкого. Было обращено внимание на отсутствие визуализации тимуса.
Ребенок в возрасте 4 месяцев был прооперирован в НПМЦ ДКК, наложен левосторонний модифицированный анастомоз Блелока. Вторым этапом хирургической коррекции сердечно-сосудистых пороков были произведены анастомозы от нисходящей аорты для обеспечения адекватного кровоснабжения правого легкого в возрасте 9 месяцев.
В ходе неоднократно проводимых гематологических исследований на пред- и постоперационных этапах стабильно выявлялась выраженная лимфопения, что вновь заставило думать о наличии у пациента синдрома иммунологической недостаточности. Минимальные показатели абсолютного и относительного количества лимфоцитов в периферической крови наблюдались в возрасте 4,5 месяца жизни и достигали 16 % (1,552 • 103/мкл).
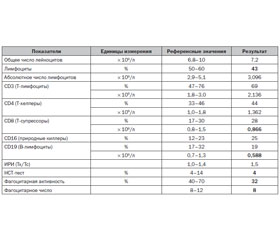
Проведенное в возрасте 6 месяцев иммунологическое исследование выявило нарушения Т-клеточного звена и показателей фагоцитоза. Отмечалось снижение относительного количества лимфоцитов, абсолютных показателей В-лимфоцитов (CD19), НСТ-теста, фагоцитарной активности и фагоцитарного числа. Абсолютные показатели Т-супрессоров (CD8) находились на нижней границе нормы (табл. 1).
/126.jpg)
В связи с подозрением на синдром Ди Джорджи (синдром делеции 22-й хромосомы на поддиске 22q11.2) было проведено молекулярно-цитогенетическое исследование ФГА-стимулированной культуры лимфоцитов периферической крови методом FISH-анализа на интерфазных ядрах с использованием Vysis TUPLE1(HIRA) Spectrum Orange/LSI ARSA Spectrum Green [Abbott]. FISH-анализ показал потерю одного красного сигнала пробы TUPLE1(HIRA), что соответствует гену, локализованному на хромосоме 22 на поддиске 22q11.2 в 39 проанализированных из 100 интерфазных ядер. Анализ контрольной пробы ARSA, которая расположена на хромосоме 22 на диске 22q13 во всех проанализированных ядрах, выявил по два сигнала (зеленый цвет). Был определен кариотип nuc ish (TUPLE1x1)[39/100], (ARSAx2)[100]. Таким образом, в 39 % случаев наблюдались клетки с делецией 22q11.2, что характерно для синдрома Ди Джорджи.
В настоящее время ребенку 2 года. Физическое развитие среднее, снижен индекс массы тела, что соответствует 1-й степени гипотрофии. Нервно-психическое развитие характеризуется некоторым отставанием речевого и сенсорного развития. У ребенка не наблюдаются выраженные признаки дисморфии лица. Можно отметить лишь относительно низкое расположение ушных раковин, их незначительную деформацию, низкий лоб. Анализ анамнестических данных позволил отметить позднюю дентацию, перенесенную инфекцию мочевыводящих путей, дисбиоз кишечника, нечастые и нетяжелые острые респираторные инфекции.
Таким образом, клинико-анамнестические критерии и результаты проведенных исследований позволили думать о наличии у ребенка неполного синдрома Ди Джорджи (синдром делеции хромосомы 22q11.2). Учитывая невысокий инфекционный индекс, а также периодически наблюдаемые нормальные показатели лимфоцитов в периферической крови, можно думать о благоприятном прогнозе для данного пациента в плане нивелирования в дальнейшем иммунологической недостаточности.
Приведенный случай демонстрирует сложность диагностики синдрома Ди Джорджи у детей, так как при неполном СДД, сопровождающемся умеренными или незначительными иммунологическими и метаболическими нарушениями, на первый план в клинической картине выходит ВПС. В наблюдаемом случае это привело к ограниченному наблюдению пациента у кардиохирургов. Несмотря на стабильное выявление лимфопении при гематологическом обследовании, пациенту не проводилось повторных исследований иммунограммы, паратиреоидного гормона, не контролировался уровень сывороточного кальция.
Необходимо акцентировать внимание врачей и родителей таких пациентов на комплексном наблюдении за ребенком педиатром, иммунологом, эндокринологом и кардиологом с проведением УЗИ тимуса, мониторированием иммунологических показателей, уровня паратгормона, кальция и фосфора.
С учетом распространенности синдрома делеции хромосомы 22q11.2 у детей с ВПС комплексный подход к обследованию пациентов с кардиологической патологией должен включать не только динамическое наблюдение узких специалистов, но и проведение иммунологической и генетической диагностики.
Тактика ведения пациента с СДД должна быть ориентирована не только на хирургическую коррекцию ВПС, но и на профилактику повторных инфекционных заболеваний и гипокальциемии, что предупреждает осложненное течение заболевания и значительно улучшает прогноз в отношении продолжительности и качества жизни ребенка.
Список литературы
1. Аллергология и иммунология: Национальное руководство / Под ред. Р.М. Хаитова, Н.И. Ильиной. — М.: ГЭОТАР-Медиа, 2009. — 656 с.
2. Белозеров Ю.М. Детская кардиология (наследственные синдромы) / Ю.М. Белозеров. — Элиста: ЗАО «РНН Джангар», 2008. — 400 с.
3. Значення цитогенетичного та молекулярно-цитогенетичних методів дослідження для верифікації діагнозу синдрому мікроделеції 22q11.2 / Н.Г. Горовенко, О.Г. Євсєєнко, Т.Е. Зерова-Любимова, Н.О. Тищенко // Лікарська справа. — 2007. — № 4. — С. 34-40.
4. Синдром Ди Джорджи: от классической триады к многообразию клинических проявлений / Н.В. Нагорная, Е.В. Бордюгова, К.В. Муравская и др. // Современная педиатрия. — 2012. — № 5(45). — С. 151-155.
5. Костюченко Л.В. Сучасні можливості діагностики та лікування синдрому Ді Джорджи в Україні / Л.В. Костюченко, Я.Ю. Романишин, А.В. Бондаренко // ПАГ. — 2011. — Т. 73, № 4(446). — С. 178-184.
6. Рекомендации по выявлению и ведению пациентов с синдромом делеции 22q11.2 / В.Г. Антоненко, Н.П. Котлукова, Ю.О. Козлова, Т.В. Золотухина // Педиатрия. — 2013. — № 4. — С. 63-68.
7. Волянська Л.А. Клінічні аспекти синдрому Ді Джорджі у дитини / Л.А. Волянська, В.В. Стеценко, О.Г. Федорчак // Актуальні питання педіатрії, акушерства та гінекології. — 2014. — № 2. — С. 59-61.
8. Practical Guidelines for Managing Patients with 22q11.2 Deletion Syndrome / Anne S. Bassett, Donna M. McDonald-McGinn et al. // The Journal of Pediatrics. — 2011. — Vol. 159, № 2. — P. 332-339.