Резюме
Мета огляду — аналіз даних літератури щодо поширеності, етіології, генетики, патогенезу синдрому Жильбера. Проведений огляд наукової літератури щодо синдрому Жильбера за ключовими словами «синдром Жильбера», «гіпербілірубінемія», «уридиндифосфатглюкуронілтрансфераза» з використанням як пошукової системи PubMed. Беручи до уваги дослідження, проведені в останні 10 років, проаналізовані тези 75 статей. Більш детально вивчено результати дослідження, висвітлені у 28 статтях.
Цель обзора — анализ данных литературы по распространенности, этиологии, генетике, патогенезе синдрома Жильбера. Проведен обзор научной литературы по синдрому Жильбера по ключевым словам «синдром Жильбера», «гипербилирубинемия», «уридиндифосфатглюкуронилтрансфераза» с использованием в качестве поисковой системы PubMed. Принимая во внимание исследования, проведенные в последние 10 лет, проанализированы положения 75 статей. Более подробно изучены результаты исследования, которые освещены в 28 статьях.
The aim of the review was the analysis of the literature about the prevalence, etiology, genetics and pathogenesis of Gilbert’s syndrome (GS). The scientific literature regarding GS with the keywords «Gilbert's syndrome», «hyperbilirubinemia», «uridine diphosphate glucuronosyltransferase (UGT-1A)» using PubMed as a search engine was reviewed. The abstracts of 75 articles, based on investigations held within the last 10 years were analyzed. The criterion for the selection of articles for the study was based on their close relevance to the topic. The results of researches covered in 10 articles and two reports were of the top interest and deep study. In medical literature GS is described under the names of different syndromes: Gilbert’s syndrome, Meulengracht’s syndrome, Gilbert — Meulengracht syndrome, Gilbert — Lereboullet syndrome, and also such as: constitutional hepatic dysfunction, familial nonhemolytic jaundice, Gilbert’s type of hyperbilirubinemia, idiopathic unconjugated hyperbilirubinemia, Crigler — Najjar hyperbilirubinemia, Arias’ type (HBLRCN, hyperbilirubinemia I). GS is a predominantly hereditary unconjugated hyperbilirubinemia associated with the reduced activity of uridine diphosphate glucuronosyltransferase (UGT-1A) in liver, which is encrypted in external resources as ICD-10 — E80.4; OMIM — 143500; DiseasesDB — 5218; MedlinePlus — 000301; eMedicinemed — 870; MeSHD — 005878. UGT-1A isoforms are found in different parts of the gastrointestinal tract (UGT1A1, UGT1A3, UGT1A4, UGT1A6), in the liver — UGT1A9, in the esophagus and stomach — UGT1A7, in the esophagus and intestines — UGT1A8, in the esophagus, bile ducts, stomach, intestines — UGT1A10, in kidneys — UGT1A19. The patients with GS have signs of disorders in all phases of metabolism of bilirubin — from its production to excretion: the lack of bilitranslocase which is responsible for the capture of bilirubin from the blood and its transport to hepatocytes, the deficit of Y- and Z-protein ligand (glutathione-S-transferase enzyme), responsible for transport of bilirubin to microsomes, the deficiency of UGT-1A, which provides the transfer of glucuronic acid to bilirubin. The prevalence of the mutant gene in European countries reaches 35–40 %, in certain ethnic groups in Africa more than 50 %, in Asian countries it is slightly lower (16–33 %). The prevalence of GS in Ukraine has not been studied. The ratio of male and female patients with GS is 3–4 : 1. The main reason of the lack of the enzyme is mutation of the gene encoding UGT1A1, however, the other factors are also responsible for the development of the syndrome and manifestation of its symptoms (male gender, additional gene mutations: c.993 (p.Q331H); *6 (c.211G > A); (nt-211, nt-686, nt-1,091 and nt-1456). The specific polymorphism in candidate genes was identified (SLCO1B3 ABCC2 and NUP153). The histological and ultrastructural features of GS include normal hepatic lobules and deposition of bile pigment granules in hepatocytes. Signs of the hepatosis are seen at later terms and serve as evidence of the evolution of the disease.
Статтю опубліковано на с. 82-86
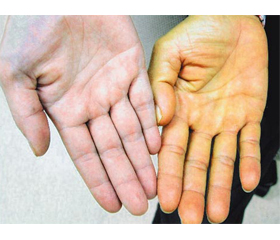
Синдром Жильбера (СЖ) — спадкова некон’югаційна гіпербілірубінемія, що пов’язана зі зниженням активності уридиндифосфатглюкуронілтрансферази (УДФ-ГТ1) у печінці, що шифрується в зовнішніх ресурсах як: МКХ-10 — E80.4; OMIM — 143500; DiseasesDB — 5218; MedlinePlus — 000301; eMedicinemed — 870; MeSHD — 005878.
Нами проведений огляд наукової літератури щодо СЖ за ключовими словами «синдром Жильбера», «гіпербілірубінемія», «уридиндифосфатглюкуронілтрансфераза» із використанням як пошукової системи PubMed. Беручи до уваги дослідження, проведені в останні 10 років, проаналізовані реферати 75 статей. Критерій для відбору статей для дослідження був заснований на їх тісному зв’язку з темою. Більш детально вивчено результати дослідження, висвітлені у 28 статтях.
Доброякісне хронічне ураження печінки вперше описали в 1900–1901 роках французькі лікарі Августин Ніколя Жильбер разом із Пьєром Леребулле як la cholemie simple familiale — проста сімейна холемія [1]. На честь Жильбера дали назву цьому синдрому, однак сучасні німецькі [2] лікарі з цим не згодні: вони вважають, що синдром вперше описав в 1939 році німецький лікар Йенс Мейленграхт [3], тому називають ураження відповідно синдромом Мейленграхта. Однак пізніше було доведено, що це різні синдроми з подібною клінічною картиною. Спільними для двох синдромів є зниження рівня білірубіну при призначенні активаторів ферментів печінки, вік маніфестації, інтермітуючий характер жовтяниці, рівень білірубіну в крові не більше 80–100 мкмоль/л за рахунок некон’югованої фракції, клінічні прояви у вигляді іктеричності шкіри та слизових, диспепсії, астенії. Але при синдромі Мейленграхта має місце тільки ізольоване зниження активності УДФ-ГТ1, а мембрана гепатоцитів на відміну від синдрому Жильбера бере активну участь у захопленні білірубіну.
У медичній літературі СЖ описується як різні синдроми: синдром Жильбера (Gilbert syndrome), синдром Мейленграхта (Meulengrachts syndrome), синдром Жильбера — Мейленграхта, (Gilbert — Meulengrachts syndrome), синдром Жильбера — Лербуйе (Gilbert — Lereboullet syndrome), а також під іншими назвами, а саме: конституційна дисфункція печінки (Constitutional hepatic dysfunctiоn), негемолітична сімейна гіпербілірубінемія (Familial nonhemolytic hyperbilirubinemia), гіпербілірубінемія, тип Жильбера (Нyperbilirubinemia, Gilbert type), ідіопатична некон’югаційна гіпербілірубінемія (Idiopathic unconjugated hyperbilirubinemia), гіпербілірубінемія Криглера — Найяра, Аріас тип, (HBLRG hyperbilirubinemia, Arias type), гіпербілірубінемія I типу (Нyperbilirubinemia I).
СЖ належить до доброякісних (функціональних) гіпербілірубінемій (пігментні гепатози) — захворювань, що пов’язані зі спадковими порушеннями обміну білірубіну (ензимопатії), які проявляються хронічною або переміжною жовтяницею без вираженої первинної зміни структури та функції печінки та без явних ознак гемолізу і холестазу [5]. До функціональних гіпербілірубінемій відносять [6]:
— синдром Криглера — Найяра 1-го і 2-го типів;
— синдром Жильбера;
— синдром Мейленграхта;
— синдром Дабіна — Джонсона;
— синдром Ротора;
— синдром Люсі — Дрісколла;
— синдром Аагенеса;
— синдром Байлера;
— первинну гіпербілірубінемію.
Найбільш поширеною доброякісною гіпербілірубінемією є СЖ, хоча в середині XX століття даний синдром вважався рідкісним станом. Поширеність мутантного гена в країнах Європи сягає 35–40 % [7], в окремих етнічних групах Африки перевищує 50 % [8], в азіатських країнах виявляється трохи рідше — 16–33 % [9]. Гомозиготами є 12 % шотландців, 16 % європейців, 12 % індійців, 8 % єгиптян і 23 % афроамериканців [10]. Поширеність СЖ в Україні не вивчалася. При СЖ співвідношення чоловіків і жінок становить 3–4 : 1. Вважається, що переважання осіб чоловічої статі пов’язано з інгібуючою дією тестостерону на УДФ-ГТ1 і утворенням більшої кількості білірубіну в чоловіків [11].
З 50-х по 80-ті роки минулого століття патогенез СЖ активно вивчався. Установлено зменшення глюкуронідації непрямого білірубіну за рахунок зниження активності УДФ-ГТ1 у печінці. У 1991 році ДНК ферменту, що кон’югує білірубін, — УДФ-ГТ1 була ідентифікована та клонована [12]. Потім було доведено, що УДФ-ГТ1 є основною трансферазою, що кон’югує білірубін у печінці людини.
Сьогодні сімейство ферментів УДФ продовжують активно вивчати. Ізоформи УДФ-ГТ1 знаходять у різних відділах шлунково-кишкового тракту (УДФ-1А1, УДФ-1А3, УДФ-1А4, УДФ-1А6; у печінці — УДФ-1А9, у стравоході та шлунку — УДФ-1А7; у стравоході та кишечнику — УДФ-1А8; у стравоході, жовчних протоках, шлунку, кишечнику — УДФ-1А10; у нирках — УДФ-1А9). Функцією ферментів сімейств УДФ-1А є кон’югація як ендогенних метаболітів, гормонів, нейротрансмітерів, так і екзогенних (різних ксенобіотиків, канцерогенів, лікарських засобів) [12].
У 2000-х роках був відкритий ген, що кодує УДФ-ГТ1 (UGT1A1), і принцип його роботи. Став відомий основний поліморфізм гена УДФ-ГТ1, що призводить до зниження активності ферменту [13]. Окрім того, виявлено, що різні ізоферменти УДФ-ГТ1 людини є продуктами одного гена, розташованого на хромосомі 2q37.1 [14]. Найбільш поширеним генетичним дефектом є зміна на промоторній ділянці гена в ділянці тимін-аденіну (ТА). Наявність хоча б однієї алелі з інсерцією (7ТА) призводить до зниження експресії гена до 20 % щодо норми, що сприяє зниженню функціональної активності ферменту на 30 % і кон’югації білірубіну в гепатоцитах на 80 % щодо норми. Це призводить до зниження функціональної активності ферменту і, таким чином, збільшує ризик реалізації синдрому.
У 94 % при СЖ пошкоджуються ферменти сім’ї глікозилтрансфераз: UDP-GT1-A6 (зниження активності приблизно на 50 %) і UDP-GT1-A7 (зниження активності приблизно на 83 %). Унаслідок цього відбуваються порушення захоплення білірубіну мікросомами васкулярного полюсу печінкової клітини, порушення його транспортування глутатіон-S-трансферазою, що доставляє непрямий білірубін до мікросом гепатоцитів, а також неповноцінність мікросомального ферменту УДФ-ГТ1,
внаслідок якої відбувається кон’югація непрямого білірубіну з глюкуроновою та іншими кислотами. Через усі ці порушення при СЖ ще й активність УДФ-ГТ1 зменшується на 70–75 % [15].
Однак останнім часом отримані дані, що переконливо свідчать про гетерогенність СЖ, оскільки у хворих із цим захворюванням є ознаки порушення практично всіх етапів обміну білірубіну — від його продукції до виведення з організму [16]:
— недостатність білітранслокази, що відповідає за захоплення білірубіну з крові і його транспорт у гепатоцит;
— дефіцит Y- і Z-протеїнів-лігандів (ферменту глутатіон-S-трансферази), що відповідають за перенесення білірубіну до мікросом;
— дефіцит УДФ-ГТ1, що забезпечує перенесення глюкуронової кислоти до білірубіну.
СЖ успадковується за аутосомно-рецесивним типом із відносно низькою пенетрантністю і мінливим проявом дефекту (хворі гетерозиготні за одним мутантним геном).
Генетичний маркер СЖ — визначення кількості ТА-повторів у промоторній ділянці гена UGT1A1 (UDP-glycosyltransferase 1 family, polypeptide A1 gene). Нормальний генотип — А(ТА)6ТАА/А(ТА)6ТАА. Вирізняють різні варіанти: гомозиготні (ТА)7/(ТА)7 — динуклеотидна вставка (7 ТА-повторів) у гомозиготній формі, гетерозиготні (ТА)6/(ТА)7 — динуклеотидна вставка (7 ТА-повторів) у гетерозиготній формі, компаунд-гетерозиготні.
Окрім вставки додаткового ТА динуклеотиду в промоторному регіоні гена, СЖ може асоціюватися з частою мутацією Gly7Arg у кодуючій ділянці гена UGT1A1.
У гомозиготних носіїв мутації захворювання характеризується більш високим рівнем білірубіну та вираженими клінічними проявами. У гетерозиготних носіїв переважає латентна форма, що виявляється, як правило, у незначному зростанні рівня білірубіну. Поширеність генетичного дефекту UGT 1А1 у популяції значна, гомозиготне носійство становить від 5 до 10 % у різних регіонах, а гетерозиготне носійство сягає 40–45 % [17]. Ці цифри показують, що діагноз синдрому Жильбера є частим.
Останнім часом виявлено ще дві нові місенс-мутації гена UGT1A1. Обидві мутації були успадковані від кожного з двох батьків, у яких рівень білірубіну нормальний. Одна із них — c.993 (p.Q331H). Описана ще одна мутація — 211G > мутація в кодуванні экзону 1 гена UGT1A1 [18].
У дослідженні, проведеному [20], показано, що 146 педіатричних пацієнтів із 181 мали хоча б один гетерозиготний UGT1A1 функціональний варіант. Виявлені результати включали 17 нових варіантів UGT1A1, 7 рідкісних відомих і 1 рідкісний новий варіант. Налічувалося 129 осіб, які мали TA7(*28) промоутер-повтори, і 15 осіб, які мали *6(c.211G > A) варіацію. Із 104 осіб із гіпербілірубінеміями в 41 особи не ідентифіковані UGT1A1-варіанти. Це вказує на необхідність дослідження додаткових генів для встановлення генетичних причин СЖ.
Проведено проспективне дослідження [21] 97 пацієнтів із непрямою гіпербілірубінемією з генотипами [6TAA6 (TA), 7TAA7 (TA)] і кодуванням ділянки [нуклеотидів (nt) -211, nt-686, nt-1,091 і nt 1456] UGT1A1. У 36 пацієнтів (45,6 %) був виявлений СЖ із генотипом 7/7, серед них у 14 — із варіантом nt-686. 42 пацієнти (43,3 %) мали генотип 6/7, серед них у 36 було встановлено один або кілька варіантів мутацій у ділянці кодування. У пацієнтів із більш високим білірубіном він пов’язаний із більш високою ймовірністю розвитку СЖ: 60,0 % (P = 0,007) пацієнтів із рівнем білірубіну ≥ 2,5 мг/дл і лише 23,9 % пацієнтів із рівнем білірубіну < 2,5 мг/дл (Р = 0,0006). Гетерозиготні варіанти мутацій гена UGT1A141 виявлено в 41 із 61 пацієнта. Автори роблять висновок про необхідність подальших досліджень для підтвердження ролі одного гомозиготного варіанту або двох і більше гетерозиготних варіантів гена UGT1A1 як чинників розвитку непрямої гіпербілірубінемії.
Ще одне дослідження [23] включало дві групи зразків із країн Північної Європи, що вирізнялися різною концентрацією білірубіну в сироватці крові (база даних NOBIDA): 150 осіб із білірубіном > 17,5 мкмоль/л і 150 осіб із білірубіном < 17,5 мкмоль/л. Досліджувалися TA6 > TA7 варіант у UGT1A1 і 7 tag-SNPs у розширеному промоутер-районі UGT1A1 (аналіз гаплотипу), окремі поліморфізми в генах-кандидатах (SLCO1B3 ABCC2 і NUP153). Знайдено значні коефіцієнти співвідношення для високого рівня білірубіну для всіх обраних варіантів UGT1A1. Однак у ступеневій багатовимірній логістичній регресії аналіз усіх генетичних варіантів разом із віком, статтю, країною походження показав повторюваність тільки тих варіантів UGT1A1 (TA6 > TA7 і SLCO1B3 rs2117032 T > C), що пов’язані з більш високою концентрацією білірубіну.
Макроскопічно печінка при СЖ не змінена. Морфологічно при цій патології будь-яких ознак диспротеїнозу, некрозу в печінкових клітинах, як правило, немає. Однак багато авторів відзначають збільшення розмірів печінки при СЖ [24]. Так, за даними [25], збільшення розмірів печінки спостерігалося у 25 % хворих, у більшості з них вона виступала на 1–2 см з-під правого підребер’я по середньоключичній лінії, в окремих випадках — на 3–4 см, консистенція була м’яка, пальпація безболісна. Описуючи морфологічну картину печінки, зазначають накопичення дрібного золотистого і жовтувато-коричневого пігменту — ліпофусцину. Накопичення ліпофусцину нерідко поєднується з дрібнокраплинною (дрібнозернистою) жировою дистрофією. Вважають, що ці крапельки жиру, що утворюються при розпаді клітинних ультраструктур мітохондрій, у подальшому перетворюються в зерна ліпофусцину.
У деяких випадках відмічається гіперплазія гладкої ендоплазматичної мережі. При гістологічному та гістохімічному дослідженні біоптатів печінки зміни або не виявляють, або вони неспецифічні: відкладення пігменту в гепатоцитах, ожиріння, глікогеноз ядер, активація зірчастих ендотеліоцитів, в окремих випадках — білкова дистрофія гепатоцитів і фіброз портальних полів [27]. Гістологічні та ультраструктурні особливості СЖ включають нормальні печінкові часточки і відкладення гранул пігменту жовчі в гепатоцитах.
Загалом морфологічні зміни тканини печінки при СЖ свідчать про певне дистрофічне ураження гепатоцитів, що вкладається в картину легкого гепатозу. Гістологічні зміни в ранні терміни захворювання зазвичай не спостерігаються. Гепатоз виявляється в більш пізні терміни і служить свідченням еволюції хвороби. При СЖ може статися повне зникнення мікроворсинок мембрани судинного полюса печінкової клітини та виникнути інші ознаки дистрофії, такі як крихкість і згладжування цитоплазматичної мембрани гепатоцитів, зверненої до простору Діссе.
Згідно з результатами динамічної гепатобілісцинтиграфії порушення поглинальної й екскреторної функції гепатоцитів виявлено в 60 % дітей із СЖ. Показники, що характеризують концентраційно-евакуаторну функцію жовчного міхура, збільшені у 2/3 дітей. Одночасно з цим визначаються функціональні порушення сфінктерного апарату жовчовивідних шляхів, що може сприяти порушенню пасажу жовчі з розвитком підпечінкового холестазу.
Розрізняють вроджений варіант синдрому Жильбера, коли клінічні прояви розвиваються у віці 12–30 років без попереднього гострого вірусного гепатиту, і синдром Жильбера, клінічні прояви якого маніфестують після перенесеного гострого вірусного гепатиту. У цьому разі має місце постгепатитна гіпербілірубінемія. До того ж вона може бути пов’язана не тільки з ініціацією клінічних проявів генетичного дефекту (з істинним синдромом Жильбера), але і з розвитком хронічного вірусного гепатиту. Тобто хворим із постгепатитною гіпербілірубінемією слід ретельно контролювати та проводити диференційну діагностику між синдромом Жильбера і хронічним вірусним гепатитом [28].
Таким чином, протягом 100 років має місце еволюція уявлень про даний діагноз — від ізольованого симптому до генодіагностики. Але досі немає однозначної точки зору на оцінку стану здоров’я цих пацієнтів, проблема діагностики СЖ і його вплив на життєдіяльність організму до кінця не вивчені. Діагностика синдрому Жильбера залишається досить складною, незважаючи на розвиток діагностичних технологій. Діагноз, як правило, установлюють методом виключення із застосуванням пункційної біопсії печінки. Все це займає досить багато часу та призводить найчастіше до гіпердіагностики хронічних гепатитів, або нерідко хворі залишаються недообстеженими.
Конфлікт інтересів. Автори заявляють про відсутність конфлікту інтересів при підготовці даної статті.
Список литературы
1. Gilbert A., Lereboullet P. La cholémie simple familial. Semaine médicale. — Paris, 1901. — 21. — 241-243.
2. Strasser P.R., Midlak S.R. Сhronischer intermittierender juveniler Subikterus // World J. Gastroenterol. — 2009. — 9(18). — 2398-407.
3. Meulengracht E. Icterus intermittens juvenilis (chronischer intermittierender juveniler Subikterus). — Klinische Wochenschrift, Berlin, 1939. — 45. — 118-121.
4. Piter J., Bosma U.A. The genetic basis of the reduced expression of bilirubin UDP-glucuronosyltransferase 1 in Gilbert’s syndrome // New England Journal of Medicine. — 1995. — № 18. — S. 1171-1175. — DOI: 10.1056/NEJM199511023331802.
5. Подымова С.Д. Пигментные гепатозы: Руководство по гастроэнтерологии / Под ред. Ф.И. Комарова, А.Л. Гребенева. — М., 1995. — Т. 2. — С. 126-32.
6. Захарова И.Н., Пыков М.И., Калоева З.В. Апостериорная ценность клинических и лабораторных проявлений синдрома Жильбера у детей // Педиатр. фармакол. — 2011. — 8(4). — 101-5.
7. Biondi M.L., Turri O., Dilillo D. et al. Contribution of the TATA-Box genotype (Gilbert’s syndrome) to serum bilirubin concentrations in the Italian population // Clin. Chem. — 1999. — 45(6). — 897-8.
8. Horsfall L.J., Zeitlyn D., Tarekegn A. et al. Prevalence of clinically relevant UGT1A alleles and haplotypes in African populations // Ann. Human Genetics. — 2010. — 75. — 236-46.
9. Balram C., Sabapathy K., Fei G., Khoo K.S., Lee E.J. Genetic polymorphisms of UDP-glucuronosyltransferase in Asians: UGT1A1*28 is a common allele in Indians // Pharmacogenetics. — 2002. — 12. — 81-3.
10. Ando Y., Chida M., Nakayama K., Saka H., Kamataki T. The UGT1A1*28 allele is relatively rare in a Japanese population // Pharmacogenetics. — 1998. — 8. — 357-60.
11. Strassburg C.P., Kalthoff S., Ehmer U. Variability and function of family 1 uridine‑5’-diphosphate glucuronos-yltransferases (UGT1A) // Crit. Rev. Clin. Lab. Sci. — 2008. — 45(6). — 485-530.
12. D’Angelo R., Rinaldi C., Donato L., Nicocia G., Sidoti A. The combination of new missense mutation with [A(TA)7TAA] dinucleotide repeat in UGT1A1 gene promoter causes Gilbert's syndrome // Ann. Clin. Lab. Sci. — 2015. — 45(2). — 202-5. — PMID: 25887876.
13. Shiu T.Y., Huang H.H., Lin H.H., Shih Y.L., Chu H.C., Chang W.K., Hsieh T.Y. Restriction fragment length polymorphism effectively identifies exon 1 mutation of UGT1A1 gene in patients with Gilbert’s Syndrome // Liver. Int. — 2015 Aug. — 35(8). — 2050-6. — DOI: 10.1111/liv.12785.
14. Hu R.T., Wang N.Y., Huang M.J., Huang C.S., Chen D.S., Yang S.S. Multiple variants in UGT1A1 gene are factors to develop indirect hyper-bilirubinemia // Hepatobiliary Surg. Nutr. — 2014 Aug. — 3(4). — 194-8. — DOI: 10.3978/j.issn.2304-3881.2014.08.04.
15. Mlakar V., Mlakar S.J., Marc J., Ostanek B. Preparation of reference material for UGT1A1 (TA)n polymorphism genotyping // Clin. Chim. Acta. — 2014, Aug 5. — 435. — 24-8. — DOI: 10.1016/j.cca.2014.04.018.
16. De Vries H.S., Te Morsche R.H., Jenniskens K., Peters W.H., de Jong D.J. A functional polymorphism in UGT1A1 related to hyperbilirubinemia is associated with a decreased risk for Crohn’s disease // J. Crohn’s Colitis. — 2012. — 6(5). — 597-602.
17. Balram C., Sabapathy K., Fei G., Khoo K.S., Lee E.J. Genetic polymorphisms of UDP-glucuronosyltransferase in Asians: UG T1A1*28 is a common allele in Indians // Pharmacogenetics. — 2002. — 12. — 81-3.
18. Rasool A., Khan M.Z., Khan F.Y. Gilbert's syndrome — a concealed adversity for physicians and surgeons // J. Аyub. Med. Coll. Abbottabad. — 2015 Jul — Sep. — 27(3). — 707-10.
19. Radlović N. Hereditary hyperbilirubinemias // Srp. Arh. Celok Lek. — 2014 Mar — Apr. — 142(3–4). — 257-60.
20. Skierka J.M., Kotzer K.E., Lagerstedt S.A., O’Kane D.J., Baudhuin L.M. UGT1A1 genetic analysis as a diagnostic aid for individuals with unconjugated hyperbilirubinemia // J. Pediatr. — 2013 Jun. — 162(6). — 1146-52, 1152, e1-2. — DOI: 10.1016/j.jpeds.2012.11.042.
21. Hu R.T., Wang N.Y., Huang M.J., Huang C.S., Chen D.S., Yang S.S. Multiple variants in UGT1A1 gene are factors to develop indirect hyper-bilirubinemia // Hepatobiliary Surg. Nutr. — 2014 Aug. — 3(4). — 194-8. — DOI: 10.3978/j.issn.2304-3881.
22. Strassburg C.P. Gilbert-Meulengracht’s syndrome and pharmacogenetics: is jaundice just the tip of the iceberg? // Drug Metabolism Reviews. — 2010. — 42(1). — 168-81.
23. Kringen M.K., Piehler A.P., Grimholt R.M., Opdal M.S., Haug K.B., Urdal P. Serum bilirubin concentration in healthy adult North-Europeans is strictly controlled by the UGT1A1 TA-repeat variants // Plos one. — February, 2014. — 28(10). — 1643-7. — DOI: 10.1371/journal.pone.0090248.
24. Sticova E., Jirsa M. New insights in bilirubin metabolism and their clinical implications // World J. Gastroenterol. — 2013. — 19(38). — 6398-407.
25. Райзис А.Р., Хохлова О.Н., Никитина Т.С. Синдром Жильбера: современные воззрения, исходы и терапия // Доктор ру. — 2012. — 3. — 42-45.
26. De Vries H.S., Te Morsche R.H., Jenniskens K., Peters W.H., de Jong D.J. A functional polymorphism in UGT1A1 related to hyperbilirubinemia is associated with a decreased risk for Crohn’s disease // J. Crohns Colitis. — 2012. — 6(5). — 597-602.
27. Poynard T., Morra R., Ingiliz P., Imbert-Bismut F., Thabut D., Messous D. [at al.]. Biomarkers of liver fibrosis // Adv. Clin. Chem. — 2008. — 46. — 131-60.
28. Zhu X.Y., Liu L.X. Ultrastructure of hepatocytes in Gilbert’s syndrome patients and chronic hepatitis B patients // Zhonghua Gan Zang Bing Za Zhi. — 2013. — 21(12). — 929-33. — doi: 10.3760/cma.j.issn.1007-3418.2013.12.011.