Международный неврологический журнал 7 (93) 2017
Нейропатичні форми лізосомних хвороб накопичення в Україні
Авторы: Пічкур Н.О.(1, 2)
(1) — Центр орфанних захворювань, Національна дитяча спеціалізована лікарня «Охматдит», м. Київ, Україна
(2) — Національна медична академія післядипломної освіти імені П.Л. Шупика МОЗ України, м. Київ, Україна
Рубрики: Неврология
Разделы: Клинические исследования
Резюме
Актуальність. На сьогодні виявлені кілька тисяч спадкових захворювань, що характеризуються ураженням нервової системи. До них належать лізосомні хвороби накопичення. Група цих захворювань включає майже 60 різних нозологічних форм спадкових моногенних порушень обміну речовин. Клінічна картина лізосомних хвороб характеризується прогредієнтним перебігом, мультисистемними порушеннями (ураження кісток скелета, м’язів, серцево-судинної, дихальної систем тощо). За тяжкого перебігу цих захворювань відзначають ураження нервової системи. Нейродегенеративні захворювання у дітей зазвичай спричинені лізосомними хворобами накопичення (ЛХН). Діагностика лізосомних хвороб в Україні розпочата у 1995 р. в НДСЛ «Охматдит». Уперше в Україні проаналізовані всі випадки лізосомних хвороб накопичення, діагностовані в період 1995–2016 рр., а також їх поширеність в українській популяції. Останніми роками спектр лабораторної діагностики ЛХН суттєво розширився, накопичено унікальний досвід клінічної ідентифікації різних форм цих рідкісних захворювань, що позначається на частоті їх виявлення, дає можливість аналізувати епідеміологічні показники, розробляти ефективні заходи щодо оптимізації ранньої діагностики. Мета. Вивчення поширеності та спектра нейропатичних форм лізосомних хвороб накопичення в Україні, порівняння отриманих результатів з даними епідеміологічних досліджень, проведених в інших країнах. Матеріали та методи. Проаналізовано клінічні та лабораторні дані пацієнтів, у яких діагностовано різні форми лізосомних хвороб, у тому числі і нейропатичні, за період спостереження 1995–2016 рр. НДСЛ «Охматдит» — єдиний лікувальний заклад в Україні, де здійснюється клініко-лабораторна діагностика цих захворювань. Поширеність лізосомних хвороб розраховували як відношення загальної кількості пацієнтів із діагнозом певної ЛХН до загальної кількості дітей, народжених живими у той самий період часу, що і хворі, в яких у подальшому діагностовано лізосомні хвороби (з розрахунку на 100 000 осіб, які народилися живими). Результати. Діагноз лізосомної хвороби встановлено у 343 хворих серед 324 родин з усіх регіонів України. Найбільш численними в структурі лізосомних хвороб виявилися сфінголіпідози (52 % спостережень) і мукополісахаридози (30 %). Нейропатичний варіант перебігу діагностований у 84 (47 %) серед 179 хворих із групи сфінголіпідозів. Хворобу Гоше діагностовано у 19,4 % хворих серед усіх лізосомних хвороб. ІІ тип хвороби Гоше (гостра нейропатична форма) відзначений у 3 (5 %) пацієнтів, ІІІ тип — у 5 (8 %). Проте, за даними дослідників з інших країн, у різних популяціях частка нейропатичної форми хвороби Гоше різниться, що, ймовірно, зумовлене особливостями певної популяції. GM1- і GM2-гангліозидози діагностовано нами у 8 % спостережень, метахроматична лейкодистрофія — у 7 %. Друге місце в структурі лізосомних хвороб посідає велика гетерогенна група мукополісахаридозів — 30 % спостережень. Синдром Хантера діагностований у 34 % спостережень у структурі мукополісахаридозів, синдром Санфіліппо (найчастіше типу А) — у 24,4 %. Нейрональний цероїдліпофусциноз ІІ типу діагностований нами у 22 хворих серед 22 родин, нейрональний цероїдліпофусциноз І типу не виявлений в жодного пацієнта. Вік 85 % (n = 291) пацієнтів, у яких було встановлено діагноз лізосомного захворювання, був молодше 18 років. Вікові інтервали селективної вибірки пацієнтів для подальшої лабораторної діагностики мають неабияке значення. Певною проблемою для України є несвоєчасне виявлення лізосомних захворювань у регіонах. Висновки. Структура лізосомних хвороб накопичення, виявлених в Україні, цілком відповідає такій в інших країнах, де ці хвороби ефективно діагностують упродовж тривалого часу. Нейропатичні форми становлять 57 % у структурі лізосомних хвороб накопичення. Ця гетерогенна група захворювань, що вимагає складної поетапної клініко-лабораторної діагностики, і є однією з основних причин виникнення нейродегенеративних захворювань у дитячому віці. Дані нашого дослідження щодо поширеності нейропатичних форм ЛХН в Україні є попередніми і потребують подальшого вивчення.
Актуальность. На сегодняшний день обнаружено несколько тысяч наследственных заболеваний, характеризующихся поражением нервной системы. К ним относят лизосомные болезни накопления (ЛБН). Группа этих заболеваний включает почти 60 различных нозологических форм наследственных моногенных нарушений обмена веществ. Клиническая картина лизосомных болезней характеризуется прогредиентным течением, мультисистемными нарушениями (поражение костей скелета, мышц, сердечно-сосудистой, дыхательной систем и т.д.). При тяжелом течении этих заболеваний отмечают поражение нервной системы. Лизосомные болезни накопления являются наиболее частой причиной нейродегенеративных заболеваний у детей. Диагностика лизосомных болезней в Украине начата в 1995 году в НГСБ «Охматдет». Впервые в Украине проанализированы все диагностированные случаи лизосомных болезней накопления, которые были выявлены в период 1995–2016 гг., а также распространенность этих заболеваний в украинской популяции. В последние годы спектр лабораторной диагностики ЛБН существенно расширился, накоплен уникальный опыт клинической идентификации различных форм этих редких заболеваний, что сказывается на частоте их диагностики, позволяет анализировать эпидемиологические показатели и разработать эффективные меры по оптимизации ранней диагностики. Цель. Изучение распространенности и спектра нейропатических форм лизосомных болезней накопления в Украине, сравнение результатов с данными эпидемиологических исследований, проведенных в других странах. Материалы и методы. Проведен анализ клинических и лабораторных данных пациентов, у которых диагностированы различные формы лизосомных болезней, в том числе и нейропатические, за период наблюдения 1995–2016 гг. НГСБ «Охматдет» — единственное лечебное учреждение в Украине, в котором проводится клинико-лабораторная диагностика этих заболеваний. Распространенность лизосомных заболеваний рассчитывали как соотношение общего количества пациентов с определенным диагнозом лизосомной болезни с общим количеством живорожденных в тот самый период времени, что и пациенты, у которых в дальнейшем было диагностировано лизосомное заболевание (из расчета 1 случай на 100 000 живорожденных). Результаты. Диагноз лизосомного заболевания был установлен у 343 пациентов среди 324 семей со всех регионов Украины. Наиболее численной группой в структуре лизосомных болезней выявились сфинголипидозы (52 % случаев) и мукополисахаридозы (30 %). Нейропатические варианты диагностированы у 84 (47 %) среди 179 пациентов из группы сфинголипидозов. Болезнь Гоше диагностирована нами у 19,4 % пациентов. ІІ тип болезни Гоше (острая нейропатическая форма) выявлен у 3 (5 %) пациентов, ІІІ тип — у 5 (8 %). По данным исследователей из других стран, часть нейропатических форм болезни Гоше отличается в зависимости от популяции. GM1- и GM2-ганглиозидозы диагностированы нами в 8 % случаев, метахроматическая лейкодистрофия — в 7 %. Второе место в структуре лизосомных болезней занимают мукополисахаридозы — 30 % случаев. Синдром Хантера диагностирован в 34 % всех клинических наблюдений в структуре мукополисахаридозов, синдром Санфилиппо (чаще тип А) — в 24,4 %. Нейрональный цероидлипофусциноз ІІ типа диагностирован у 22 больных среди 22 семей, нейрональный цероидлипофусциноз І типа не выявлен ни у одного пациента. Возраст 85 % (n = 291) пациентов, у которых был установлен диагноз лизосомного заболевания, был младше 18 лет. Возрастные интервалы селективной выборки пациентов для дальнейшей лабораторной диагностики имеют важное значение. Важной проблемой для Украины стало несвоевременное выявление лизосомных заболеваний в регионах. Выводы. Структура лизосомных болезней накопления, выявленных в Украине, полностью соответствует данным, полученным в других странах, где эти болезни эффективно диагностируют на протяжении длительного периода. Нейропатические формы составляют 57 % в структуре лизосомных болезней. Эта гетерогенная группа заболеваний, требующих сложной поэтапной клинико-лабораторной диагностики, — одна из основных причин нейродегенеративных заболеваний центральной нервной системы детского возраста. Данные исследования распространенности нейропатических форм лизосомных болезней в Украине являются предварительными и требуют дальнейшего изучения.
Background. To date, several thousands of hereditary diseases have been detected, characterized by the defeat of the nervous system. These include lysosomal storage diseases (LSD). Lysosomal storage desiaeses — is a large group of monogenic metabolic disorders (about 60), inherited mainly by the autosomal recessive type; most of them — are serious diseases with progressive flow that cause severe disability and early death of the patients. LSD are characterized by accumulation of enzymes substrates or products of their incomplete degradation and segregated cytoplasmic components, etc. in different cells and tissues and it causes many disorders. Depending on the accumulated substrate nature, the following LSD are distinguished: mucopolysaccharidosis, sphingolipidosis, mucolipidosis, oligosaccharidosis, etc. Certain LSD clinical flow depends on the pathological process type and location, accumulated biochemical substrate nature, and is usually multisymptomatic. Within one nosological form various phenotypes are found: from «soft» to severe; first symptoms of the disease may be nonspecific, reminiscent of other, more common diseases. Most LSD require complex stage-by-stage clinical and laboratory diagnostics. Until recently almost all LSD considered as incurable; specific correction methods were successfully developed and introduced into medical — it has become one of the most significant achievements in a worldwide medical science of recent decades. Recently, significantly expanded range of LSD laboratory diagnostics gained unique experience in clinical identification of their various forms that significantly influenced on the disease incidence revealing, allowed to analyze epidemiological indicators, to develop effective measures for early diagnosis optimization. The purpose of the study was to study the prevalence and spectrum of neuropathic forms of lysosomal accumulation diseases in Ukraine, comparing the results with epidemiological studies conducted in other countries. Materials and methods. LSD enzyme diagnostics in Ukraine is used since 1995: today it is possible to determine the activity of 25 enzymes in homozygous leukocytes, blood plasma, chorionic biopsy, skin fibroblasts cells culture that allowed to verify 30 different diseases. Each LSD has its own biochemical characteristics. The laboratory diagnostics success depends on a clear definition of a clinical problem. This approach is a key to correct diagnosis, further clinical and laboratory monitoring during LSD treatment. Data of the complex clinical and laboratory examination of patients with different forms of LSD provided in the National Specializes Children Hospital «Okhmatdit» in the period 1995–2016 have been analyzed. For LSD laboratory diagnostics we selected patients with multisymptomatic clinical picture which were grouped on the basis of certain organs and systems disorders; for each group phase-based research protocols were used. Results. The diagnosis of lysosomal disease was established in 343 patients from 324 families from all regions of Ukraine. The most numerous group in the structure of lysosomal diseases revealed sphingolipidosis (52 % of cases) and mucopolysaccharidosis (30 %). Neuropathic variants were diagnosed in 84 (47 %) of 179 patients from the sphingolipidosis group. We diagnosed Gaucher disease in 19.4 % of patients. The second type of Gauchers disease (acute neuropathic form) was detected in 3 (5 %) patients, type III in 5 (8 %). The most numerous in LSD structure were sphingolipidosis (52 %) and mucopolysaccharidosis (30 %); a significant place took GM1-gangliosidosis (8 %) and metachromatic leukodystrophy (7 %). According to our research, LSD structure in Ukraine is similar to other European countries, but in the same time the incidence of Gaucher and Fabry disease, metachromatic leukodystrophy, mucopolysaccharidosis type I was lower, probably due to LSD significant clinical polymorphism and a large number of adult patients. Conclusions. In the future we are planning to study LSD prevalence in Ukraine, to analyze the mistakes in their late diagnostics in order to develop and implement in the medical practice effective early diagnostic algorithms for certain forms of LSD, for which there are effective methods of treatment.
Ключевые слова
лізосомні хвороби накопичення; нейропатичні форми; сфінголіпідози; мукополісахаридози; нейрональний цероїдліпофусциноз; метахроматична лейкодистрофія; біохімічна, молекулярно-генетична діагностика
лизосомные болезни накопления; нейропатические формы; сфинголипидозы; мукополисахаридозы; нейрональный цероидлипофусциноз; метахроматическая лейкодистрофия; биохимическая, молекулярно-генетическая диагностика
lysosomal storage diseases; neuropathic forms; sphingolipidosis; mucopolysaccharidosis; neuronal ceroid lipofuscinosis; metachromatic leukodystrophy; biochemical, molecular and genetic diagnosis
Робота є фрагментом НДР «Визначення генетичних основ ризику розвитку патологічних станів на різних етапах онтогенезу» (2014–2018) за № держреєстрації 0114U002215, кафедра медичної та лабораторної генетики, Національна медична академія післядипломної освіти імені П.Л. Шупика МОЗ України, м. Київ, Украї–на.
Вступ
На сьогодні виявлено кілька тисяч спадкових захворювань, що характеризуються ураженням нервової системи [1]. Зокрема, до них належать метаболічні захворювання, спричинені генетично детермінованою дисфункцією ферментів і некаталітичних протеїнів, необхідних для функціонування лізосом, та відомі як лізосомні хвороби накопичення (ЛХН) [2, 3].
ЛХН — велика (майже 60 різних нозологічних форм) група спадкових моногенних порушень обміну речовин [4], що характеризуються накопиченням різноманітних сполук, зокрема, субстратів ферментів або продуктів їх неповної деградації, сегрегованих цитоплазматичних компонентів, у лізосомах різних клітин і тканин. Майже всі ЛХН спричинені зниженням активності ферментів лізосом внаслідок мутацій у кодуючому гені відповідного ферменту або захисного білка, білка-активатора, а також системи посттрансляційної модифікації білка [5]. Проте деякі ЛХН спричинені дисфункцією лізосом, яка виникає внаслідок мутації в генах, що кодують компоненти мембран або протеїнів, тісно пов’язані з системою лізосом. Кількість ферментів лізосом і некаталітичних протеїнів, описаних у науковій літературі, постійно збільшується, отже, цілком імовірним є виявлення ще більшої кількості дефектів системи лізосом [6].
ЛХН — рідкісні хронічні захворювання з прогредієнтним перебігом, численними різноманітними симптомами, для яких характерні мультисистемні порушення (ураження кісток скелета, м’язів, серцево-судинної, дихальної систем тощо). За тяжкого перебігу ЛХН відзначають ураження нервової системи. Нейродегенеративні захворювання у дітей зазвичай спричинені ЛХН [7]. Відзначають різні варіанти перебігу ЛХН: від «м’якої» форми до тяжкої, крім того, перші ознаки захворювання нерідко є неспецифічними, через це у таких хворих нерідко помилково діагностують інші, більш поширені захворювання. Більшість ЛХН спричиняють ранню тяжку інвалідизацію та смерть хворих [5]. ЛХН класифікують відповідно до природи патологічного субстрату, який накопичується у лізосомах: мукополісахаридози (МПС), ліпідози, сфінголіпідози, олігосахаридози [8–10].
Нещодавно практично всі ЛХН вважали некурабельними. Проте за останні десятиліття з’явилася реальна можливість ефективного лікування метаболічних порушень при ЛХН шляхом застосування ферментозамісної, шаперонної, субстрат-редукційної терапії [11–13]. Активно розробляються методи генної терапії, зокрема, при нейропатичних формах ЛХН [14].
Важливу роль у створенні ефективної системи надання медичної допомоги пацієнтам при ЛХН відіграють два аспекти: рання й точна клініко-лабораторна діагностика та вивчення поширеності ЛХН у популяції.
Поширеність ЛХН вивчали в країнах із високорозвинутою системою надання медичної допомоги пацієнтам при спадкових метаболічних захворюваннях, в яких упроваджені методи лабораторної діагностики орфанних захворювань [15–17].
За даними дослідників, поширеність ЛХН у різних популяціях становить від 7,6 до 25 на 100 000 населення [18–20].
Діагностика ЛХН в Україні розпочата в 1995 р. у НДСЛ «Охматдит». Останніми роками спектр лабораторної діагностики ЛХН суттєво розширився, накопичений унікальний досвід клінічної ідентифікації різних форм цих рідкісних захворювань, що позначається на частоті їх виявлення, дозволяє аналізувати епідеміологічні показники, розробляти ефективні заходи щодо оптимізації ранньої діагностики ЛХН.
Мета дослідження: вивчення поширеності та спектра нейропатичних форм ЛХН в Україні, порівняння отриманих результатів із даними епідеміологічних досліджень, проведених в інших країнах.
Матеріали та методи
Проаналізовано клінічні та лабораторні дані пацієнтів, у яких діагностовано різні форми ЛХН, за період спостереження 1995–2016 р. НДСЛ «Охматдит» — єдиний лікувальний заклад в Україні, в якому здійснюється клініко-лабораторна діагностика ЛХН. До 2008 р. верифікацію діагнозу здійснювали у Медико-генетичному центрі лікарні, з 2008 р. і дотепер — в Центрі орфанних захворювань; створена єдина в Україні база пацієнтів, які страждають від ЛХН. Для проведення лабораторної діагностики при припущенні про наявність ЛХН відбирали пацієнтів із полісиндромною клінічною картиною. Були сформовані селективні групи, в які включали хворих залежно від наявного комплексу синдромів ураження певних органів і систем, для кожної з яких розроб–лені поетапні протоколи лабораторного дослідження. Так, хворих при ураженні центральної нервової системи (регрес психомоторних функцій, епілепсія, резистентна до медикаментозного лікування, лейкодистрофія, атрофічні зміни головного мозку за даними магнітно-резонансної томографії (МРТ)) направляли на обстеження з приводу гангліозидозів, хвороби Краббе, метахроматичної лейкодистрофії (МЛД), нейронального цероїдліпофусцинозу (НЦЛ). При переважанні у клінічній картині гематологічного синдрому (анемія, тромбоцитопенія, гепатоспленомегалія) хворих обстежували на наявність сфінгомієлінозів (хвороба Гоше, хвороба Німана — Піка). За наявності ознак гаргоїлізму (низький зріст, ураження кісткової системи) хворих обстежували на мукополісахаридоз. За наявності нейропатичних форм ЛХН необхідна комплексна оцінка загального, неврологічного статусу пацієнта, МРТ головного мозку.
Лабораторне дослідження у хворих і членів їх родин проводили в лабораторії медичної генетики НДСЛ «Охматдит» з використанням біохімічних і молекулярно-генетичних методів.
Біохімічне дослідження при припущенні про наявність ЛХН починали з визначення вмісту глікозаміногліканів (ГАГ) у сечі або ЦПХ-тесту, хроматографії у тонкому шарі олігосахаридів і ГАГ [21–23]. У подальшому визначали дефекти певних ферментів, зокрема, лізосомних гідролаз із використанням синтетичних високоспецифічних субстратів за стандартними методиками [24–27]. Зазвичай для ферментної діагностики використовували гомогенат лейкоцитів, інколи визначали активність ферментів у плазмі крові, при припущенні про наявність хвороби Помпе — у сухій плямі крові [18, 28].
Поширеність ЛХН визначали як відношення загальної кількості пацієнтів із діагнозом певної ЛХН до загальної кількості дітей, народжених живими у той самий період часу, що і хворі, в яких у подальшому діагностовано ЛХН (з розрахунку на 100 000 осіб, які народилися живими). Період народження — це період часу між роком народження найстаршого і наймолодшого пацієнтів. Дані щодо народжуваності взяті з офіційного сайту Державної статистичної служби України (ukrstat.gov.ua).
При деяких специфічних ЛХН пацієнтів розподіляли на підгрупи з вираженими клінічними фенотипами (зокрема, при МЛД, хворобі Гоше). Поширеність певної ЛХН визначали як суму поширеності всіх її підгруп [18, 29–34].
При визначенні поширеності в популяції Х-зчеплених захворювань, зокрема, хвороби Фабрі, МПС ІІ типу, вивчали специфічні клінічні прояви у хворих обох статей [20, 35].
Результати та обговорення
Діагноз ЛХН встановлений у 343 хворих серед 324 родин з усіх регіонів України.
Етапність діагностики ЛХН включає встановлення попереднього клінічного діагнозу на підставі виявлення комплексу характерних симптомів з подальшим лабораторним підтвердженням наявності захворювання. Лабораторна діагностика ЛХН також є поетапною, застосовують різні клінічні, морфологічні, біохімічні та молекулярно-генетичні дослідження. ЛХН характеризуються клінічним поліморфізмом і генетичною гетерогенністю. Крім того, при деяких ЛХН клінічна картина подібна до такої при інших спадкових і набутих захворюваннях. Чітке визначення клінічних критеріїв для формування певних селективних груп на етапі встановлення попереднього клінічного діагнозу має важливе значення для вибору протоколу подальшого лабораторного дослідження та успішності біохімічного дослідження. Такий підхід є запорукою встановлення правильного діагнозу ЛХН і подальшого клініко-лабораторного моніторингу стану хворих під час лікування.
Ферментні дослідження при припущенні про наявність ЛХН в Україні проводяться з 1995 р. На сьогодні можливе визначення активності 25 ферментів лізосом у гомогенаті лейкоцитів, плазмі крові, біоптаті хоріона, культурі фібробластів шкіри, що дозволяє верифікувати 30 нозологічних форм ЛХН.
Виявлені нами спостереження ЛХН подані в табл. 1.
У 18 спостереженнях в одній родині діагностовано дві ЛХН. При визначенні частки окремих захворювань у структурі ЛХН ураховували кількість родин, обтяжених певним захворюванням, незалежно від кількості уражених сибсів.
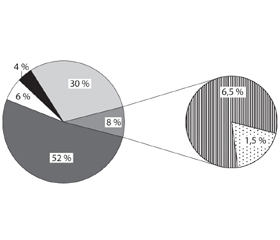
Найбільш численними в структурі ЛХН виявилися сфінголіпідози (52 % спостережень) і МПС (30 %) (рис. 1). Отримані нами результати цілком збігаються з даними іноземних дослідників [20].
Нейропатичні форми ЛХН зазвичай виявляють при ліпідозах, зокрема, сфінголіпідозах (МЛД, гангліозидозах (GM1-гангліозидоз (Тея — Сакса), GM2-гангліозидоз (Сандхоффа)), хворобах Краббе, Німана — Піка типу С, Гоше ІІ і ІІІ типів [2]. Нейропатичний варіант перебігу сфінголіпідозу діагностований у 84 (47 %) серед 179 хворих (табл. 1, рис. 2).
Хвороба Гоше діагностована нами у 19,4 % родин, обтяжених ЛХН. Хвороба Гоше І типу характеризується ураженням внутрішніх органів і кісток скелета; ІІ тип (гострий) — це фатальна нейропатична форма захворювання, перші ознаки якої виявляють у перші місяці життя дитини, зазвичай хворі живуть не довше 2 років; ІІІ тип — проміжний (хронічний): симптоми помірного дегенеративного ураження нервової системи та внутрішніх органів, перші ознаки захворювання виявляють у більш дорослому віці, тривалість життя таких хворих близько 30 років [36, 37].
Характерними симптомами нейропатичного варіанта хвороби Гоше є окуломоторна апраксія, супрануклеарна офтальмоплегія, екстрапірамідні порушення, генералізовані міоклонічні судоми, мозочкова атаксія, затримка психічного та мовного розвитку. Найчастіше першою ознакою захворювання є горизонтальний супрануклеарний парез погляду [38, 39].
Слід зауважити, що нейропатичну форму хвороби Гоше виявляють вкрай рідко: у 6 % хворих на хворобу Гоше, при цьому у 5 % спостережень діагностують хронічну нейропатичну форму захворювання і лише в 1 % — гостру [40].
Хворобу Гоше діагностовано нами у 19,4 % хворих на ЛХН. ІІ тип захворювання (гостра нейронопатична форма) відзначений у 3 (5 %) пацієнтів, ІІІ тип — у 5 (8 %). Проте, за даними дослідників з інших країн, у різних популяціях частка нейропатичної форми хвороби Гоше різниться, що, ймовірно, зумовлене особливостями певної популяції [41, 42].
GM1- і GM2-гангліозидози діагностовано нами у 8 % спостережень, МЛД — у 7 % (табл. 1).
Друге місце в структурі ЛХН посідає велика гетерогенна група МПС — 30 % спостережень. Синдром Хантера діагностований у 34 % спостережень у структурі МПС, синдром Санфіліппо (найчастіше типу А) — у 24,4 % (рис. 3).
Для МПС характерні виражені нейродегенеративні форми захворювання. МПС І типу (хвороба Гурлера) характеризується тяжким клінічним перебігом; його виявлено у 7 хворих. МПС ІІ типу (хвороба Хантера) також має нейропатичний варіант перебігу, що характеризується порушенням поведінки, прогресуючим зниженням інтелекту, епілептичним синдромом, у термінальній стадії захворювання може виникнути спастичний тетрапарез. Нейропатичний варіант МПС ІІ типу виявлений у 10 пацієнтів серед 9 родин.
На відміну від МПС І і ІІ типів МПС ІІІ типу (хвороба Санфіліппо) є суто нейродегенеративним захворюванням, що об’єднує гетерогенну групу нозологій з автосомно-рецесивним типом успадкування, спричинених зниженням активності одного з 4 ферментів, залучених у процес послідовного розщеплення гепарансульфату [43–45]. Залежно від біохімічного дефекту виділяють 4 підтипи МПС ІІІ: А, B, C і D. У більшості (майже 70 % спостережень) хворих діагностований МПС ІІІ А (табл. 1) [19]. Результати нашого дослідження відповідають даним інших авторів [18, 19, 46–48].
Для МПС ІV типу характерний винятково «скелетний» фенотип, для МПС VІ типу — «вісцеральний», без ознак нейродегенеративного процесу [10].
МПС спричинені порушенням інтралізосомального транспорту. Це група ЛХН, які зазвичай виникають дуже рано (інколи — під час внутрішньоутробного розвитку), характеризуються широким спектром клінічних проявів і тяжким перебігом (муколіпідоз ІІ α/β-типу і ІІІ α/β-типу) або пізнім дебютом з «м’якою» клінічною картиною (муколіпідоз ІІІ γ-типу), що нагадує ревматоїдний артрит [10]. Прогредієнтний перебіг характерний для муколіпідозу ІІ α/β-типу. Муколіпідоз ІІ α/β-типу діагностований у 14 хворих, ІІІ α/β-типу — у 5.
Глікопротеїнози (манозидози α і β, сіалідози, фукозидоз та ін.) — гетерогенна група ЛХН, спричинених порушенням метаболізму глікопротеїнів. За даними літератури, в Чеській Республіці глікопротеїнози становлять 6,4 % в структурі ЛХН, їх виявляють в 1 на 100 000 новонароджених (так само, як у Нідерландах і Австралії) [10, 20]. Глікопротеїнози виявлені нами у 4 % спостережень, що, ймовірно, зумовлене особливостями української популяції або тим, що ця патологія є надзвичайно рідкісною, що створює певні труднощі при її діагностиці.
НЦЛ об’єднує 11 гетерогенних фатальних нейродегенеративних нозологій. Зокрема, хвороба Баттена характеризується прогредієнтним перебігом, втратою хворим усіх набутих навичок, виникненням епілепсії, резистентної до медикаментозного лікування. Вважають, що саме НЦЛ найчастіше спричиняють нейродегенеративні стани у хворих дитячого віку, однак точно визначити їх поширеність на сьогодні неможливо. За даними British Pediatric Surveillance Unit, щороку у Великій Британії у 10–12 осіб діагностують різні типи НЦЛ, при цьому майже у 50 % спостережень — НЦЛ ІІ типу (www.ucl.ac.uk/ncl). НЦЛ І і ІІ типів в Центрі орфанних захворювань діагностують з 2007 р.; НЦЛ ІІ типу діагностований нами у 22 хворих серед 22 родин, НЦЛ І типу не виявлений в жодного пацієнта. Діагностика інших типів НЦЛ потребує подальшої роботи для впровадження у вітчизняну медичну практику.
За результатами нашого дослідження, структура ЛХН в Україні подібна до такої в інших країнах Європи. Проте, поширеність хвороби Гоше, хвороби Фабрі, МЛД і МПС І типу виявилася нижчою, ніж в інших країнах світу, ймовірно, через значний клінічний поліморфізм цих захворювань і, як наслідок, існування великої кількості їх «дорослих» форм. Такі показники зумовили необхідність визначення поширеності певних ЛХН з урахуванням великого періоду часу, протягом якого народилися хворі, включені у наше дослідження. Так, при хворобі Гоше найстарший пацієнт у нашому дослідженні був 1942 року народження, наймолодший — 2014 року народження. Проте ЛХН зазвичай діагностували у хворих молодше 18 років (у 291 (85 %) пацієнтів).
Вікові інтервали селективної вибірки пацієнтів для подальшої лабораторної діагностики мають неабияке значення. Певною проблемою для України є несвоєчасне виявлення ЛХН у регіонах, несвоєчасне направлення пацієнтів за припущення про наявність ЛХН для проведення специфічної діагностики, особливо дорослого населення. Крім того, цілу низку ЛХН в Україні почали діагностувати нещодавно, що не дає можливості вірогідно визначити їх поширеність у популяції. Саме тому отримані нами результати не дозволяють зробити остаточні висновки щодо вірогідної частоти виявлення ЛХН серед населення України. В подальшому планується вивчення більш широкого спектра ЛХН в Україні, аналіз помилок їх пізньої діагностики з метою розробки та впровадження в медичну практику ефективних алгоритмів ранньої діагностики деяких форм ЛХН, для яких розроблені ефективні методи лікування.
Висновки
Структура ЛХН, виявлених в Україні, цілком відповідає такій в інших країнах, у яких ЛХН ефективно діагностують упродовж тривалого часу.
Нейропатичні форми (57 % у структурі ЛХН) — гетерогенна група захворювань, які вимагають складної поетапної клініко-лабораторної діагностики й є однією з основних причин виникнення нейродегенеративних захворювань у дитячому віці.
Дані нашого дослідження щодо поширеності нейропатичних форм ЛХН в Україні є попередніми і потребують подальшого вивчення.
Вдячність. Автор висловлює щиру подяку за спів–працю під час діагностики ЛХН лікарям медико-генетичних центрів України, Центру орфанних захворювань, лікарям-лаборантам Медико-генетичного центру НДСЛ «Охматдит», співробітникам кафедри медичної та лабораторної генетики Національної медичної академії післядипломної освіти імені П.Л. Шупика МОЗ України.
Конфлікт інтересів. Автор заявляє про відсутність конфлікту інтересів при підготовці даної статті.
Список литературы
1. Korf B.R. New approaches to molecular diagnosis / B.R. Korf, H.L. Rehm // JAMA. — 2013. — Vol. 309, N 14. — P. 1511-1521.
2. Cox T.M. The cellular pathology of lysosomal disease / T.M. Cox, M.B. Cachon-Gonzalez // J. Pathol. — 2012. — Vol. 226, N 2. — P. 241-254.
3. Platt F.M. The cell biology of disease: lysosomal storage disorders: the cellular impact of lysosomal dysfunction / F.M. Platt, B. Boland, A.C. van der Spoel // J. Cell. Biol. — 2012. — Vol. 199, N 5. — P. 723-734.
4. Coutinho M.F. From rare to common and back again: 60 years of lysosomal dysfunction / M.F. Coutinho, S. Alves // Mol. Genet. Metab. — 2015. — Vol. 117, N 2. — P. 53-65.
5. Jalanko A. Neuronal ceroid lipofuscinoses / A. Jalanko, T. Braulke // Biochim. Biophys. Acta. — 2009. — Vol. 1793, N 4. — P. 697-709.
6. Berkovic S.F., Dibbens L.M., Oshlack A., Silver J.D., Kate–relos M., Vears D.F. [et al.]. Array-based gene discovery with three unrelated subjects shows SCARB2/LIMP-2 deficiency causes myoclonus epilepsy and glomerulosclerosis // Am. J. Hum. Genet. — 2008. — Vol. 82, N 3. — P. 673-684.
7. Verity C., Winstone A.M., Stellitano L., Will R., Nicoll A. The epi–demiology of progressive intellectual and neurological deterioration in childhood // Arch. Dis. Child. — 2010. — Vol. 95, N 5. — P. 361-364.
8. Zlotogora J., Furman-Shaharabani Y., Harris A., Barth M.L., von Figura K., Gieselmann V. A single origin for the most frequent mutation causing late infantile metachromatic leukodystrophy // J. Med. Genet. — 1994. — Vol. 31, N 6. — P. 672-674.
9. Schultz M.L., Tecedor L., Chang M., Davidson B.L. Clarifying lysosomal storage diseases // Trends Neurosci. — 2011. — Vol. 34, N 8. — P. 401-410.
10. Lysosomal storage disorders. A practical guide / Ed. by A. Mehta, B. Winchester. — Hoboken: Wiley-Blackwell, 2012. — 208 p.
11. Wenger D.A. Insights into the diagnosis and treatment of lysosomal storage diseases / D.A. Wenger, S. Coppola; Shu-Ling Liu // Arch Neurol. — 2003. — Vol. 60. — P. 322-328.
12. Macualey S.L. Combination therapies for lysosomal storage diseases:a complex answer to a simple problem / S.L. Macualey // Ped. Endocrinol. Rev. — 2016. — Vol. 13, suppl. 1. — P. 639-645.
13. Grabowski G.A. Enzyme therapy for lysosomal storage dise–ase: principles, practice, and prospects / G.A. Grabowski, R.J. Hopkin // Ann. Rev. Genom. Hum. Genet. — 2003. — Vol. 4. — P. 403-436.
14. Yew N.S. Cheng gene therapy for lysosomal storage disorders / N.S. Yew, H.S. Cheng // Ped. Endocrinol. Rev. — 2013. — Vol. 11, suppl. 1. — P. 99-105.
15. Dionisi-Vici C., Rizzo C., Burlina A.B., Caruso U., Sabetta G., Uziel G. [et al.]. Inborn errors of metabolism in the Italian pediatric population: a national retrospective survey // J. Pediatr. — 2002. — Vol. 140, N 3. — P. 321-327.
16. Pinto R., Caseiro C., Lemos M., Lopes L., Fontes A., Ribeiro H. Prevalence of lysosomal storage diseases in Portugal // Eur. J. Hum. Genet. — 2004. — Vol. 12, N 2. — P. 87-92.
17. Guo Y., He W., Boer A.M., Wevers R.A., de Bruijn A.M., Groener J.E. [et al.]. Elevated plasma chitotriosidase activity in various lisosomal storage disorders // J. Inher. Metab. Dis. — 1995. — Vol. 18. — P. 717-722.
18. Poorthuis B.J., Wevers R.A., Kleijer W.J., Groener J.E., de Jong J.G., van Weely S. [et al.]. The frequency of lysosomal storage diseases in the Netherlands // Hum. Genet. — 1999. — Vol. 105. — P. 151-156.
19. Miekle P.J. Prevalence of lysosomal storage disorders / P.J. Meikle, J.J. Hopwood, W.F. Clague // JAMA. — 1999. — Vol. 281. — P. 249-254.
20. Poupětová H., Ledvinová J., Berná L., Dvořáková L., Kožich V., Elleder M. The birth prevalence of lysosomal storage disorders in the Czech Republic: comparison with data in different populations // J. Inherit. Metab. Dis. — 2010. — Vol. 33, N 4. — P. 387-396.
21. Мицик Н.Й. Диференціація норми та патології методом селективного біохімічного скринінгу лізосомних хвороб накопичення, що супроводжуються підвищеною екскрецією олігосахаридів / Н.Й. Мицик, Н.В. Ольхович, Н.Г. Горовенко // Укр. біохім. журн. — 2015. — Т. 87, № 3. — P. 107-115.
22. Трофімова Н.С. Використання скринуючих біохімічних досліджень для ранньої діагностики мукополісахаридозів /
Н.С. Трофімова, Н.В. Ольхович, Н.Г. Горовенко // Вісн. проблем біології і медицини. — 2015. — Т. 2(123), вип. 3. — С. 245-250.
23. Мицик Н.Й. Селективний біохімічний скринінг лізосомних хвороб накопичення методом тонкошарової хроматографії олігосахаридів / Н.Й. Mицик, Н.В. Ольхович, Н.Г. Горовенко // Вісн. проблем біології і медицини. — 2016. — Т. 126, № 1. — С. 222-227.
24. Горовенко Н.Г. Молекулярно-генетичний скринінг мажорних мутацій в гені АСА у пацієнтів з метахроматичною лейкодистрофією / Н.Г. Горовенко, Н.В. Ольхович, Н.О. Пічкур // Цитология и генетика. — 2002. — Т. 36, № 5. — С. 43-49.
25. Wenger D.A. Screening for lysosomal disorders / D.A. Wenger, C. Williams // Techniques in diagnostics of human biochemical gene–tics. — N.Y.: Wiley-Liss, 1991. — P. 587-619.
26. Hartree E.F. Determination of protein: a modification of the Lowry method that gives a linear photometric response / E.F. Hartree // Annal. Biochem. — 1972. — Vol. 48, N 4. — P. 422-427.
27. Мицик Н.Й. Особливості оцінки активності β-галактозидази в діагностиці лізосомних хвороб накопичення серед населення України / Н.Й. Mицик, Н.В. Ольхович, Н.Г. Горовенко // Вісн. проблем біології і медицини. — 2016. — Т. 133, № 4. — С. 208-213.
28. Lukacs Z., Nieves Cobos P., Mengel E., Hartung R., Beck M., Deschauer M. [et al.]. Diagnostic efficacy of the fluorometric determination of enzyme activity for Pompe disease from dried blood specimens compared with lymphocytes-possibility fornewborn screening // J. Inherit. Metab. Dis. — 2010. — Vol. 33. — P. 43-50.
29. Горовенко Н.Г. Використання бioxiмічниx методів для визначення гетерозиготного носійства метахроматичної лейкодистрофії / Н.Г. Горовенко, Н.В. Ольхович // Проблеми екологічної та медичної генетики i клінічної імунології. — 2003. — № 3(49). — С. 35-42.
30. Ольхович Н.В., Грищенко О.М., Пічкур Н.О., Недобой А.М., Трофімова Н.С., Іванова Т.П. [та ін.]. Клініко-лабораторні показники ефективності ферментозамісної терапії хвороби Гоше в Україні // Лік. справа. — 2011. — № 1–2. — С. 95-104.
31. Mistry P.K. The glucocerebrosidase locus in Gaucher’s disease: molecular analysis of lysosomal enzyme / P.K. Mistry, T.M. Cox // J. Med. Genet. — 1993. — Vol. 30. — P. 889-894.
32. Polten A., Fluharty A.L., Fluharty C.B., Kappler J., von Figura K., Gieselmann V. Molecular basis of different forms of metachromatic leukodystrophy // N. Engl. J. Med. — 1991. — Vol. 324, N 1. — P. 18-22.
33. Lugowska A., Amaral O., Berger J., Berna L., Bosshard N.U., Chabas A. [et al.]. Mutations c.459+1G>A and p.P426L in the ARSA gene: prevalence in metachromatic leukodystrophy patients from European countries // Mol. Genet. Metab. — 2005. — Vol. 86. — P. 353-359.
34. Olkhovich N.V., Takamura N., Pichkur N.A., Gorovenko N.G., Aoyagi K., Yamashita S. Novel mutations in arylsulfatase A gene in three Ukrainian families with metachromatic leukodystrophy //
Mol. Genet. Metabol. — 2003. — Vol. 80, N 3. — Р. 360-363.
35. Трофімова Н.С. Оптимізація біохімічної та молекулярно-генетичної діагностики мукополісахаридозу І типу в Україні / Н.С. Трофімова, Н.В. Ольхович, Н.Г. Горовенко // Досягнення біології та медицини. — 2014. — № 1(23). — С. 61-65.
36. Tylki-Szymanska A., Millat G., Maire I., Czartoryska B. Types I and III Gaucher Disease in Poland: Incidence of the Most Common Mutations and Phenotypic Manifestations // Eur. J. Hum. Genet. — 1996. — Vol. 4. — P. 334-337.
37. Jakobkiewicz-Benecka J., Gabig-Ciminska M., Banecka-Majkutewicz Z., Banecki B., Wegrzyn A., Wegrzyn G. Factors and processes modulating phenotypes in neuropathic lysosomal storage diseases // Metab. Brain Dis. — 2014. — Vol. 29. — P. 1-8.
38. Beutler E., Grabowski G.A. Gaucher disease. In: Scri–ver C.R., Beaudet A.C., Sly W.S., Valle D. et al. The metabolic and molecular basis of inherited disease. — New York: McGraw-Hill Book Co, 1995. — 7th ed. — Vol. 2. — P. 2641-70.
39. Davies E.H., Mengel E., Tylki-Szymanska A., Kleinotiene G., Reinke J., Vellodi A. Four-year follow-up of chronic neuronopathic Gaucher disease in Europeans using a modified severity scoring tool // J. Inherit. Metab. Dis. — 2011. — Vol. 34, N 5. — P. 1053-1059.
40. Charrow J., Andersson H.C., Kaplan P., Kolodny E.H., Mistry P., Pastores G. [et al.]. The Gaucher Registry: demographics and disease characteristics of 1698 patients with Gaucher disease // Arch. Intern. Med. — 2000. — Vol. 160, N 18. — P. 2835-2843.
41. Tylki-Szymańska A., Vellodi A., El-Beshlawy A., Cole J.A., Kolodny E. Neuronopathic Gaucher disease: demographic and clinical features of 131 patients enrolled in the International Collaborative Gaucher Group Neurological Outcomes Subregistry // J. Inherit. Metab. Dis. — 2010. — Vol. 33, N 4. — P. 339-346.
42. Горовенко Н.Г., Ольхович Н.В., Недобой А.М., Пічкур Н.О. Визначення частоти мажорних мутацій в гені GBA у пацієнтів з хворобою Гоше в Україні // Цитологія і генетика. — 2007. — Т. 41, № 4. — С. 41-48.
43. Weber B., van de Kamp J.J.P., Kleijer W.J., Guo X.H., Blanch L., van Diggelen O.P. [et al.]. Identification of a common mutation (R245H) in Sanfilippo A patients from the Netherlands // J. Inherit. Metab. Dis. — 1998. — Vol. 21. — P. 416-422.
44. Trofimova N.S. Specificities of Sanfilippo A syndrome laboratory diagnostics / N.S. Trofimova, N.V. Olkhovych, N.G. Gorovenko // Biopolymers and Cell. — 2014. — Vol. 30, N 5. — P. 388-393.
45. Scriver C.R., Beaudet A.L., Sly W.S., Valle D. The metabolic and molecular basis of inherited diseases.— N.Y.: McGraw-Hill, 2001.
46. Bunge S., Ince H., Steglich C., Kleijer W.J., Beck M., Zaremba J. [et al.]. Identification of 16 sulfamidase gene mutations including the common R74C in patients with mucopolysaccharidosis type IIIA (Sanfilippo A) // Hum. Mutat. — 1997. — Vol. 10. — P. 479-485.
47. Moore D., Connock M.J., Wraith E., Lavery C. The prevalence of and survival in Mucopolysaccharidosis I: Hurler, Hurler-Scheie and Scheie syndromes in the UK // Orphanet. J. Rare Dis. — 2008. — Vol. 3. — P. 24.
48. Baehner F., Schmiedeskamp C., Krummenauer F., Miebach E., Bajbouj M., Whybra C. [et al.]. Cumulative incidence rates of the mucopolysaccharidoses in Germany // J. Inherit. Metab. Dis. — 2005. — Vol. 28, N 6. — P. 1011-1017.

/49-1.jpg)
/50-1.jpg)
/50-2.jpg)