Відомо, що імуноглобуліни виконують роль антитіл і синтезуються плазматичними клітинами завдяки антигенному стимулу та хелперному сигналу. Основна функція IgA в організмі людини — це захист мукозального епітелію за рахунок специфічного розпізнавання антигенів і гаптенів та взаємодії з іншими складовими секрету. Мономерні одиниці імуноглобулінів складаються з ідентичних двох важких та двох легких ланцюгів, які утримуються разом дисульфідними ковалентними й нековалентними зв’язками [1]. У сироватці крові IgA (IgA1, мономер) становить 15 % від усіх імуноглобулінів, його нормальна концентрація залежить від віку (табл. 1), а період напіврозпаду становить 5–6 діб. Сироватковий імуноглобулін А здатен елімінувати мікроби завдяки активації фагоцитозу й комплементу альтернативним шляхом.
/118-1.jpg )
Секреторний IgA (IgA2, димер) відрізняється наявністю додаткового секреторного компонента (S), що синтезується епітеліальними клітинами слизових оболонок та приєднується до молекули IgA при її проходженні через епітеліальні клітини. S-компонент обумовлює стійкість молекули імуноглобуліну до дії протеолітичних ферментів. IgA2 виділяється зі слини, сльози, бронхіального секрету, тонкого кишечника, він частково блокує процеси адгезії вірусів до слизової оболонки, у значній концентрації — інгібує їх прикріплення до клітинної стінки, а в невеликій концентрації — припиняє реплікацію вірусу, не змінюючи адгезію. Термінальна манозозв’язувальна ділянка важкого ланцюга молекули IgA здатна розпізнавати манозоспецифічні лектини, наявні на ворсинках І типу, що забезпечує неспецифічний антиадгезивний ефект широкого спектра бактерій. Взаємодія IgA з фагоцитами та лімфоцитами в слизовій оболонці та лимфоїдних органах периферії, що експресують рецептори до Fc-фрагмента імуноглобуліну, активує антитілозалежну клітинну цитотоксичність проти багатьох бактерій. Отже, головна роль димерного IgA пов’язана з його заданістю зв’язувати харчові антигени, алергени, інфекційні агенти, що розташовані в просвіті кишечника. Окрім того, фізіологічна роль секреторного IgA включає інгібіцію активності запалення, що запобігає автоімунній активації [2].
Селективний дефіцит IgA є найбільш поширеним первинним імунодефіцитом і характеризується ізольованим дефіцитом IgA (< 0,07 г/л, що дорівнює < 7 мг/дл) зі зазвичай нормальним рівнем IgM та IgG у пацієнтів, старших за 4 роки, коли інші етіологічні фактори зниження його рівня виключені. Вважають, що в дітей, молодших за 4 роки, зниження рівня імуноглобулінів може бути фізіологічним. У частини пацієнтів з дефіцитом IgA можуть бути знижені рівні субкласів IgG.
Критерії SIgAD за European Society for Immunodeficiencies (ESID) подані нижче [3]. Селективний IgA має місце в дитини, старшої за 4 роки, коли:
— діагностовано підвищену чутливість до захворювань пазух носа та/або легень або автоімунні захворювання в дитини та/або члена сім’ї;
— рівень сироваткового IgA < 0,07 г/л, а загальна кількість IgG і IgM у сироватці нормальна;
— виключені інші причини гіпогаммаглобулінемії;
— рівень IgG антитіл до всіх щеплень нормальний;
— виключений дефект Т-клітин.
Рівень сироваткового IgA < 0,07 г/л — основний критерій SIgAD.
Можлива присутність секторного IgA в системах слизової в дітей з SIgAD, особливо в осіб із безсимптомним носійством. За даними Міжнародного союзу імунологічних товариств (International Union of Immunological Societies, IUIS) щодо первинних імунодефіцитних захворювань, дефіцит IgA має два підтипи, які включають дефіцит IgA, пов’язаний із дефіцитом підкласу IgG, та селективний дефіцит IgA, і це неодноразово наголошувалось у період з 2007 по 2014 р. [3]. Однак в останній перегляд класифікації IUIS у 2015 році дефіцит IgA включений не був [3]. Отже, європейська класифікація SIgAD поки базується на звіті 2014 року, який включає розподіл дефіциту IgA на два підтипи.
У 2015 році групою науковців із Швеції та Ірану запропонована клінічна класифікація фенотипів SIgAD, що буде присутня в наступному перегляді IUIS [4]:
Фенотип SIgAD із безсимптомним перебігом.
Фенотип SIgAD із легким перебігом захворювань пазух носа та легень.
Фенотип SIgAD із проявами алергії.
Фенотип SIgAD з автоімунним синдромом.
Фенотип SIgAD із тяжким перебігом.
Клінічні фенотипи SIgAD із тяжким перебігом і проявами алергії мають тенденцію до маніфестації в дитинстві, тоді як у середньому віці діагностують фенотип SIgAD із легким перебігом захворювань пазух носа й легень та автоімунний фенотип. Тяжкі алергічні та автоімунні фенотипи не підлягають перекласифікації у фенотип SIgAD із безсимптомним перебігом; водночас до безсимптомного перебігу з віком часто додаються інші фенотипи.
Епідеміологія селективного дефіциту IgA
Селективний дефіцит IgA є одним із найбільш поширених захворювань первинного імунодефіциту. Захворюваність на SIgAD залежить від етнічного походження — 1 : 651 в Ірані; 1 : 143, 1 : 163 в Іспанії; 1 : 252, 1 : 875 в Англії й 1 : 965 у Бразилії. Жителі Кавказу, Африки та Середнього Сходу мають найбільшу частоту захворювання (від 1 : 100 до 1 : 1000). У мешканців Японії та Китаю IgA дефіцит становить від 1 : 1600 до 1 : 1900. Загальна частота може бути більш високою, оскільки в більшості осіб SIgAD перебігає безсимптомно, а програми скринінгу дефіциту IgA немає. Сімейні дослідження пацієнтів із SIgAD як пробандів свідчать про те, що родичі першого ступеня в 7,5 % мають SIgAD [3–6].
Патогенез селективного дефіциту IgA
Точний молекулярний дефект, який обумовлює селективний IgA дефіцит, поки що невідомий. Оскільки SIgAD має неоднорідний характер, то можна припустити, що етіологія асоціюється з внутрішнім дефектом В-клітинних лімфоцитів, Т-клітинними аномаліями та порушеннями в мережах цитокінів. На рис. 2 поданий нормальний розвиток В-клітин та етап, на якому синтез IgA може блокуватися при селективному дефіциті IgA [7].
Виявлено, що пацієнти із SIgAD мають дефект перемикання класу імуноглобулінів, термінальної диференціації IgA позитивних плазмабластів у секреторні клітини або меншу тривалість виживання IgA-секретуючих плазматичних клітин за рахунок схильності до швидшого апоптозу В-клітин, що містять IgA [8]. Ig-несучі В-клітини є позитивними щодо синтезу IgM та IgD, проте негативні до IgA. При SIgAD описана генетично порушена перебудова перемикача (switch, S) із Sμ на Sα в периферичних В-клітинах: виявлено низьку експресію як секретованих, так і мембранних форм повної мРНК у B-клітинах з IgA при SIgAD. Окрім того, при SIgAD доведений дефект у продукції TGF-b, IL-4, IL-6, –IL-7, IL-10 та IL-21, що може бути пов’язаний зі зменшенням або порушенням Т-клітинної активності хелперів у деяких пацієнтів із SIgAD [3, 10].
Цитогенетичні дефекти та мутації, пов’язані із селективним дефіцитом IgA
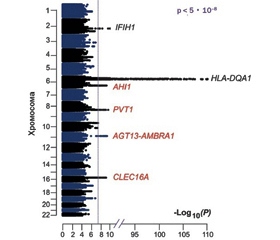
Хромосомні аномалії та цитогенетичні дефекти часто виявляються в пацієнтів з IgAD, включаючи моносомію за 4p, трисомію за 8-ю, 10p, 21-ю хромосомою, транслокацію 10q до 4p, делецію 17p11.2, 18q-синдром та синдром делеції 22q11.2. Окрім того виявляються моногенні мутації. Метааналіз генотипу 6487 добровольців, закінчений у 2016 році, виявив 4 нові значущі локуси хромосом, асоційовані з дефіцитом IgA. Окрім IFIH1 локусів на 2-й хромосомі та HLA-DQA1 на 6-й хромосомі, з дефіцитом IgA пов’язані PVT1 (8-ма хромосома), ATG13-AMBRA1 (11-та хромосома), AHI1 (6-та хромосома) та CLEC16A (16-та хромосома) (рис. 3). Ці дані дозволяють припустити, що дефіциту IgA сприяє складна мережа генетичних ефектів, включаючи гени, які, як відомо, впливають на фізіологію вироблення IgA.
Клінічна маніфестація селективного дефіциту IgA
Більшість пацієнтів з дефіцитом IgA не мають клінічних ознак або захворювання переносять легко. У невеликої групи пацієнтів захворювання перебігає тяжко. У даний час незрозуміло, чому деякі люди з дефіцитом IgA майже не хворіють, а інші хворіють тяжко. Також точно не відомо, у якого відсотка осіб із дефіцитом IgA розвиватимуться ускладнення, оцінки становлять від 25 до 50 %. Деякі пацієнти з дефіцитом IgA також мають дуже низький рівень підкласів IgG (як правило, IgG2 і/або IgG3–4). Це може бути поясненням того, чому деякі пацієнти з дефіцитом IgA більш чутливі до інфекції, ніж інші, але не для всіх хворих [3].
Як визначалось вище, незважаючи на те, що більшість людей із SIgAD — безсимптомні носії, для деяких хворих характерні легеневі інфекції, алергії, автоімунні захворювання, захворювання шлунково-кишкового тракту та злоякісні новоутворення.
Легеневі хвороби є єдиною або домінуючою нозологією під час діагностики SIgAD (40–90 % від загальної кількості пацієнтів). Більшість інфекцій викликані позаклітинними інкапсульованими бактеріями (наприклад, Haemophilus influenzae, Streptococcus pneumoniae) і бувають лише у формі інфекції верхніх дихальних шляхів або більш тяжких хвороб (бронхоектази або облітеруючий бронхіоліт). У дітей, які частіше мають дефіцит підкласів IgG, особливо IgG2 та IgG3, зазвичай хвороба перебігає більш тяжко [3]. Рекомендовано визначати рівень IgA в сироватці крові в усіх дітей із рецидивними захворюваннями пазух носа та легень, у хворих на тяжкі легеневі інфекції бажано додатково аналізувати субкласи IgG (рівень доказовості А) [3]. Пацієнти із SIgAD мають компенсаторне збільшення В-клітин, що несуть IgM, через відсутність IgA.
Алергічні захворювання можуть бути першим і/або єдиним клінічним проявом у деяких пацієнтів із SIgAD (25–50 % від загальної кількості пацієнтів). З IgAD пов’язані алергічний кон’юнктивіт, риніт, кропив’янка, екзема, харчова алергія та астма. Дійсно, секреторний IgA допомагає запобігти поглинанню алергенів у кров, відіграючи значну роль у запобіганні алергії. Пацієнти із SIgAD мають підвищений ризик анафілаксії, коли вони отримують продукти крові, що містять деяку кількість IgA. Це, як вважають, пов’язано з IgG (або, можливо, IgE) анти-IgA антитілами, які можуть бути знайдені в деяких людей, дефіцитних за IgA [3].
Поширеність автоімунних захворювань у пацієнтів із SIgAD коливається від 5 до 30 %. Ідіопатична тромбоцитопенічна пурпура, хвороба Грейвса, автоімунна гемолітична анемія, цукровий діабет 1-го типу, ревматоїдний артрит, тиреоїдит, системний червоний вовчак та целіакія зустрічаються в пацієнтів із SIgAD [3]. Дефіцит IgA (у тому числі секреторного) пов’язаний з легким проникненням антигенів у слизову. Молекулярна мімікрія та перехресна реакція з автоантигенами можуть спричинити утворення автореактивних антитіл [3]. Виникає патологічна регуляція Т-клітин, особливо в CD4 + CD25 + Foxp3 + регуляторних T-клітинах (Treg), що обумовлює зниження імунної толерантності. Дослідники вважають, що існує складна асоціація між гаплотипами лейкоцитарного антигену людини (HLA) HLA-A1, HLA-B8, HLA-DR3 та HLA-DQ2 і розвитком автоімунних захворювань у дітей з SIgAD. Крім того, було показано, що сироватки пацієнтів із SIgAD часто містять автоантитіла до тиреоглобуліну, еритроцитів, мікросомальних антигенів щитоподібної залози, базальної мембрани, клітин гладкої мускулатури, клітин підшлункової залози, ядерних білків, кардіоліпіну, колагену людини та клітин надниркової залози, навіть за відсутності клінічних автоімунних проявів. Значна частина осіб із SIgAD має сироваткові антитіла до IgA, які можуть викликати реакції при інфузії препаратів крові [3].
Частота розвитку розладів шлунково-кишкового тракту в пацієнтів SIgAD невисока. IgM може компенсувати відсутність IgA в кишечнику шляхом його транспортування зі слизової оболонки в просвіт кишечника. Проте повідомляється про зв’язок між SIgAD та целіакією, лямбліозом, вузликовою лімфоїдною гіперплазією, виразковим колітом, хворобою Крона, злоякісною анемією та аденокарциномою шлунка та кишечника [3, 11].
Целіакія є більш поширеним захворюванням у дітей із SIgAD (частота дефіциту IgA у хворих на целіакію становить десь від 2 до 3 %). Секреторний IgA може зв’язуватися з деякими білками (наприклад, трансглутаміназою, гліадином і проламіном) у шлунково-кишковому тракті, а його відсутність може призводити до дисфункції розпізнавання цих антигенів. Крім того, зв’язок між целіакією та SIgAD може мати генетичну основу, зокрема деякі спільні HLA-гаплотипи, такі як HLA-A1, HLA-Cw7, HLA-B8, HLA-DR3 та HLA-DQ2. Гістопатологія целіакії при SIgAD відрізняється відсутністю IgA-секретуючих плазматичних клітин у зразках біоптатів кишечника [11, 12]. Оскільки целіакія найчастіше виявляється за наявністю IgA-антитіл до вищезгаданих білків (наприклад, трансглутамінази, гліадину та проламіну), у дітей із підозрою на SIgAD пропонується ідентифікувати IgG проти деамідованих пептидів гліадину (deamidated gliadin peptide antibody) через високу специфічність для надійної діагностики целіакії в пацієнтів із SIgAD (рівень доказовості А) [11]. Отже, результати тестів на основі IgA при целіакії можуть бути хибнонегативними, а результати тестів на основі IgG слід вважати цінними та клінічно значимими.
Можлива тривала персиситенція Giardia lamblia у дітей з SIgAD за рахунок недосконалості слизового бар’єру [3, 11]. Giardia lamblia можуть приєднуватися до епітелію й проліферувати. Цисти лямблії утворюють трофозоїти, що колонізують тонкий кишечник і викликають здуття живота, спазми та водянисту діарею. Діагноз ґрунтується на визначені цист або трофозоїтів лямблії в калі або аспіраті з дванадцятипалої кишки.
Злоякісні новоутворення при SIgAD виявляються рідко. У звіті 2010 року описуються 63 ізраїльські дитини із SIgAD, які спостерігалися протягом 10 років, серед них злоякісні захворювання діагностовано в трьох осіб (4,8 %) [13].
Лабораторні методи діагностики SIgAD у дітей [3]
1. Визначення рівня IgA в сироватці крові рекомендовано проводити всім дітям із підозрою на SIgAD (пацієнти із хронічними захворюваннями пазух носа та/або легень, алергічними хворобами, рецидивним лямбліозом, ідіопатичною тромбоцитопенічною пурпурою, хворобою Грейвса, автоімунною гемолітичною анемією, цукровим діабетом 1-го типу, ревматоїдним артритом, тиреоїдитом, системним червоним вовчаком та целіакією) (рівень доказовості А).
2. Дітям із SIgAD рекомендовано оцінювати рівень субкласів IgG2 та IgG3 у сироватці крові (рівень доказовості А).
3. У дітей з анафілактичними реакціями на препарати крові в анамнезі, автоімунними захворюваннями (ідіопатична тромбоцитопенічна пурпура, хвороба Грейвса, автоімунна гемолітична анемія, цукровий діабет 1-го типу, ревматоїдний артрит, тиреоїдит, системний червоний вовчак та целіакія) аналізуються антитіла до IgА (рівень доказовості А).
4. Пацієнтам із рівнем IgA < 7 мг/дл бажано досліджувати мутації локусів IFIH1 на 2-й хромосомі, HLA-DQA1 (6-та хромосома), PVT1 (8-ма хромосома), ATG13-AMBRA1 (11-та хромосома), AHI1 –(6-та хромосома) та CLEC16A (16-та хромосома) для уточнення діагнозу (рівень доказовості В).
Ведення пацієнтів із SIgAD
Патогенетичної терапії SIgAD не існує. У деяких осіб рівень IgA без лікування поступово стає нормальним. Часто в підлітковому віці може бути діагностований загальний варіабельний імунодефіцит (common variable immunodeficiency) [12].
Моніторинг пацієнтів із SIgAD здійснюється різними методами, враховується освіта, лікування алергічних або автоімунних захворювань, використання тривалих курсів або навіть профілактичне використання антибіотиків, застосування полівалентних пневмококових вакцин та внутрішньовенної або підшкірної замісної терапії імуноглобулінами.
Поінформованість пацієнта та освіта мають першорядне значення, особливо для запобігання потенційній анафілактичній реакції на переливання крові та/або її продукту. Щодо цього пацієнтам з SIgAD слід рекомендувати носити медичний браслет (рівень доказовості В) [3]. Рекомендується, щоб усі пацієнти, навіть із безсимптомним фенотипом, контролювали рівень IgA в сироватці крові кожні 4–6 місяців (рівень доказовості В).
У дітей із реакціями на інфузію препаратів крові рекомендовано проводити скринінговий тест на антитіла до IgA (рівень доказовості А) для запобігання рецидивуючим реакціям залежно від необхідного продукту крові. Для цих пацієнтів препарати крові повинні готуватися індивідуально або слід обирати відмиті еритроцити для лікування анемії. Усі продукти крові слід вживати з обережністю, а персонал повинен бути готовим до лікування потенційної анафілактичної реакції (рівень доказовості А).
Лікування алергічних та автоімунних захворювань
Лікування алергії та автоімунних захворювань, пов’язаних із SIgAD, відбувається за загальними принципами (рівень доказовості А). Головним питанням у менеджменті автоімунних розладів, пов’язаних із SIgAD, є рання діагностика захворювання та визначення антитіл до IgA.
Профілактична антибактеріальна терапія
У пацієнтів із хронічними захворюваннями пазух носа або легень слід почати профілактичну специфічну антибактеріальну терапію, особливо в зимові місяці (рівень доказовості А). На жаль, не завжди можливо ідентифікувати відповідні мікроорганізми та їх чутливість до антибіотиків; отже, може знадобитися використання антибіотиків широкого спектра дії або сульфаніламідів [3].
Для пацієнтів із хронічними захворюваннями пазух носа або легень пропонується використовувати додаткову імунізацію пневмококової вакциною (рівень доказовості А) [3]. Пацієнтів зі зниженою здатністю до продукції антиполісахаридних антитіл слід імунізувати кон’югатами з полісахаридами-білками, такими як Haemophilus influenzae типу b (Hib) з дифтерійним-правцевим анатоксином. Кон’югований протеїн дозволяє продукувати антитіла проти Hib, хоча зазвичай потрібні дві або три дози. Було продемонстровано, що призначення пневмококової вакцини в пацієнтів із дефіцитом IgG2 та IgG3 призвело до виробітки антитіл до кон’югованої пневмококової вакцини з подальшим зниженням частоти інфекцій [14].
Внутрішньовенна або підшкірна замісна терапія імуноглобулінами
Пацієнтам, яким проведена пневмококова вакцинація та профілактична антибактеріальна терапія, які мають хронічні захворювання пазух носа та/або легень та можуть потребувати введення внутрішньовенного імуноглобуліну, в зимові місяці рекомендована внутрішньовенна або підшкірна замісна терапія імуноглобулінами. У більшості практичних рекомендацій початкова доза IgG становить 400–600 мг/кг/місяць для досягнення рівня сироваткового рівня IgG 600–800 мг/дл [15]. Замісну терапію імуноглобуліном можна застосовувати у болюсних дозах внутрішньовенно кожні 21–28 днів, або та ж сама доза може бути розділена на щоденні. Щотижневі або щотижневі дози для підшкірного введенні розраховуються за конверсією 1 : 1. Загалом заміна імуноглобуліну повинна проводитись обережно препаратом із вмістом IgA < 10 мг/мл, що забезпечує безпечність терапії [15].
Прогноз SIgAD
Прогноз SIgAD в основному залежить від фенотипу. Зафіксовано рідкісні випадки спонтанного одужання, особливо в молодих пацієнтів. Рідко селективний дефіцит IgA може прогресувати до загального варіабельного імунодефіциту [17].
Отже, SIgAD є найчастішим первинним імунодефіцитом із поки що недоведеною етіологією. Цитогенетичні дефекти лежать в основі даного захворювання, а світовий досвід діагностики та лікування селективного IgA дефіциту може бути основою для впровадження такого в амбулаторній і стаціонарній педіатричній практиці України.
Конфлікт інтересів. Автори заявляють про відсутність конфлікту інтересів при підготовці даної статті.
Список литературы
1. Chernyshova L.I. Immunological Basis of Immunization / L.I. Chernyshova, A.P. Volokha, F.I. Lapiy // Здоров’я дитини. — 2014. — № 52. — P. 188-192.
2. Иммуноглобулин А (IgA) [Електронний ресурс] / Eurolab. Украина. — Режим доступу: http://www.eurolab.ua/services/271/
3. Yazdani R. Selective IgA Deficiency: Epidemiology, Pathoge–nesis, Clinical Phenotype, Diagnosis, Prognosis and Management / R. Yazdani, G. Azizi, H. Abolhassani // Scandinavian journal of immunology. — 2017. — № 85(1). — P. 3-12.
4. Yazdani R. Clinical phenotype classification for selective immunoglobulin A deficiency / R. Yazdani, A. Latif, F. Tabassomi et al. // Expert Rev. Clin. Immunol. — 2015. — № 11(11). — P. 1245-1254.
5. Kumar P. Immunoglobulin A deficiency in celiac disease in the United States / Kumar Pallav, Hua Xu, D.A. Leffle // Journal of Gastroenterology and Hepatology. — 2016. — № 31(1). — P. 133-137.
6. Bronson P.G. Common variants at PVT1, ATG13-AMBRA1, AHI1 and CLEC16A are associated with selective IgA deficiency / P.G. Bronson, D. Chang, T. Bhangale, M.F. Seldin // Nat. Genet. — 2016. — № 48(11). — P. 1425-1429.
7. Mertin S. What you need to know about IgA deficiency: a case study / S. Mertin, I. Thomson // J. Am. Assoc. Nurse Pract. — 2014. — № 26. — P. 268-272.
8. Cipe F. B-cell subsets in patients with transient hypogammaglobulinemia of infancy, partial IgA deficiency, and selective IgM deficiency / F. Cipe, F. Doðu, D. Güloðlu et al. // J. Investig. Allergol. Clin. Immunol. — 2013. — № 23. — P. 94-100.
9. Maruyama S. Immunoglobulin A deficiency following treatment with lamotrigine / Y Okamoto, M. Toyoshima, R. Hanaya, Y. Kawano // Brain Dev. — 2016. — № 38(10). — P. 947-949.
10. Yazdani R. Role of apoptosis in common variable immunodeficiency and selective immunoglobulin A deficiency / R. Yazdani, M. Fatholahi, M. Ganjalikhani-Hakemi et al. // Mol. Immunol. — 2016. — № 71. — P. 1-9.
11. Azizi V. Approach to the Management of Autoimmunity in Primary Immunodeficiency / V. Azizi, M. Tavakol // Scandinavian journal of immunology. — 2017. — № 85(1). — P. 13-29.
12. Jolles S. When to initiate immunoglobulin replacement the–rapy (IGRT) in antibody deficiency: a practical approach / S. Jolles, H. Chapel, J. Litzman // Clinical and Experimental Immunology. — 2017. — № 188(3). — P. 333-341.
13. Shkalim V. Selective IgA deficiency in children in Israel / V. Shkalim, Y. Monselize, N. Segal et al. // J. Clin. Immunol. — 2010. — № 30. — P. 761-765.
14. Bonilla F.A. Practice parameter for the diagnosis and management of primary immunodeficiency / F.A. Bonilla, D.A. Khan, Z.K. Ballas et al. // J. Allergy Clin. Immunol. — 2017. — № 136. — P. 1186-1205.
15. Milito C. Adequate patient’s outcome achieved with short immunoglobulin replacement intervals in severe antibody deficiencies / C. Mi–lito, F. Pulvirenti, A.M. Pesce et al. // J. Clin. Immunol. — 2014. — № 34. — P. 813-819.
16. Berger M. Bioavailability of IgG administered by the subcutaneous route / M. Berger, S. Jolles, J.S. Orange, J.W. Sleasman // J. Clin. Immunol. — 2013. — № 33. — P. 984-990.
17. Cheraghi T. Prediction of the evolution of common variable immunodeficiency: HLA typing for patients with selective IgA deficiency / T. Cheraghi, A. Aghamohammadi, B. Mirminachi et al. // J. Investig. Allergol. Clin. Immunol. — 2014. — № 24. — P. 198.

/119-1.jpg )
/120-1.jpg )