
Журнал «Почки» Том 7, №2, 2018
Вернуться к номеру
Маркери порушення фібрилогенезу у дітей з різними варіантами перебігу пієлонефриту
Авторы: Лук’яненко Н.С.(1, 2), Іськів М.Ю.(1), Кенс К.А.(2), Макух Г.В.(1)
(1) — ДУ «Інститут спадкової патології НАМН України», м. Львів, Україна
(2) — Львівський національний медичний університет ім. Данила Галицького, м. Львів, Україна
Рубрики: Нефрология
Разделы: Справочник специалиста
Версия для печати
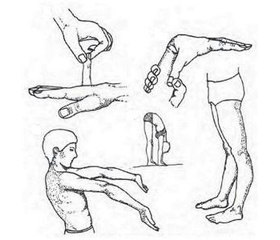
Актуальність. Останніми роками клінічний перебіг пієлонефриту у дітей характеризується збільшенням кількості латентних та «стертих» форм захворювання, підвищенням резистентності до традиційної антибактеріальної терапії, що призводить до хронізації і рецидивування патологічного процесу в нирках. При цьому в структурі нефропатій у дітей переважають захворювання вродженого та спадкового генезу, а також захворювання, пов’язані зі спадковою схильністю, що мають прихований початок та торпідний перебіг. Особливої уваги заслуговують діти з проявами дисплазії сполучної тканини. Мета дослідження: пошук можливих клініко-лабораторних маркерів, асоційованих із порушенням фібрилогенезу в дітей з різними варіантами перебігу пієлонефриту, та з’ясування ролі порушення фібрилогенезу у тяжкості перебігу пієлонефриту у дітей. Матеріали та методи. Обстежено 60 дітей з пієлонефритом від 3 до 18 років. За результатами катамнестичного спостереження вони були поділені на дві групи: І — 30 дітей з пієлонефритом, в катамнезі яких діагностувалось 3 і більше епізоди рецидиву пієлонефриту впродовж року, ІІ — 30 дітей з пієлонефритом, в яких упродовж року не відмічалось рецидивів захворювання. Контрольну групу становили 42 соматично здорові дитини того ж віку. Усім дітям проводилося рутинне комплексне клініко-лабораторне обстеження та встановлювались клініко-лабораторні маркери порушення фібрилогенезу (фенотипові ознаки недиференційованої дисплазії сполучної тканини, оксипролін в сечі). Проводилось молекулярно-генетичне тестування локусів AA та AG поліморфного локусу RS 605143 гена COL4A1. Результати. У дітей з рецидивуючим перебігом пієлонефриту вірогідно частіше відмічались фенотипові ознаки недиференційованої дисплазії сполучної тканини порівняно з даними дітей, в яких спостерігався тільки один епізод пієлонефриту протягом року, а саме: гіпермобільність суглобів (у 53 % дітей проти 6,67 % у групі порівняння, в контролі — 4,76 %), астенічна тілобудова (56,67 % проти 26,67 %, в контролі — 16,67 %), порушення зору (86,67 % проти 36,67 %, в контролі — 7,14 %), деформація грудної клітки (43,33 % проти 10 %, в контролі — 4,76 %), сколіотична постава (53,33 % проти 13,33 %, в контролі — 4,76 %), арахнодактилія та схильність до кровотеч відмічались лише у дітей І групи (23,33 та 6,67 % відповідно, в контролі не відмічалось). Визначення рівня оксипроліну у сечі в дітей з рецидивуючим пієлонефритом свідчить про посилення розпаду та екскреції продуктів обміну колагену в дитячому організмі у 97 % обстежених, що значно перевищує частоту виділення оксипроліну з сечею у дітей з гострим перебігом пієлонефриту (10 %), та вказує на виражене порушення катаболізму колагену в дітей, схильних до рецидивування пієлонефриту. Встановлена вірогідно вища частота «дикого» генотипу АА — rs605143 гена колагену COL4A1 у дітей із рецидивуючим перебігом пієлонефриту порівняно з даними осіб загальнопопуляційної контрольної групи (21,4 % проти 4,8 %, p < 0,05). Наявність у дитини генотипу АА поліморфного локусу rs605143 гена COL4A1 у п’ять разів збільшує ризик розвитку рецидивування хронічного пієлонефриту (відношення шансів 5,105, 95% довірчий інтервал 0,12–0,87). Висновки. З метою прогнозування у дітей генетично детермінованої схильності до рецидивування пієлонефриту рекомендується проведення молекулярно-генетичного тестування генотипів AA та AG поліморфного локусу rs605143 гена колагену COL4A1.
Актуальность. В последние годы клиническое течение пиелонефрита у детей характеризуется увеличением количества латентных и «стертых» форм заболевания, повышением резистентности к традиционной антибактериальной терапии, что приводит к хронизации и рецидивированию патологического процесса в почках. При этом в структуре нефропатий у детей преобладают заболевания врожденного и наследственного генеза, а также заболевания, связанные с наследственной предрасположенностью, имеющие скрытое начало и торпидное течение. Особого внимания заслуживают дети с проявлениями дисплазии соединительной ткани. Цель исследования: поиск возможных клинико-лабораторных маркеров, ассоциированных с нарушением фибриллогенеза у детей с различными вариантами течения пиелонефрита, и выяснение роли нарушения фибриллогенеза в тяжести течения пиелонефрита у детей. Материалы и методы. Обследовано 60 детей с пиелонефритом от 3 до 18 лет. По результатам катамнестического наблюдения они были разделены на две группы: I — 30 детей с пиелонефритом, в катамнезе которых диагностировалось 3 и более эпизода рецидива пиелонефрита в течение года, II — 30 детей с пиелонефритом, у которых в течение года не отмечалось рецидивов заболевания. Контрольную группу составили 42 соматически здоровых ребенка того же возраста. Всем детям проводилось рутинное комплексное клинико-лабораторное обследование и устанавливались клинико-лабораторные маркеры нарушения фибриллогенеза (фенотипические признаки недифференцированной дисплазии соединительной ткани, оксипролин в моче). Проводилось молекулярно-генетическое тестирование локусов AA и AG полиморфного локуса RS 605 143 гена COL4A1. Результаты. У детей с рецидивирующим течением пиелонефрита достоверно чаще отмечались фенотипические признаки недифференцированной дисплазии соединительной ткани по сравнению с данными детей, у которых наблюдался только один эпизод пиелонефрита в течение года, а именно: гипермобильность суставов (у 53 % детей против 6,67 % в группе сравнения, в контроле — 4,76 %), астеническое телосложение (56,67 % против 26,67 %, в контроле — 16,67 %), нарушения зрения (86,67 % против 36,67 %, в контроле — 7,14 %), деформация грудной клетки (43,33 % против 10 %, в контроле — 4,76 %), сколиотическая осанка (53,33 % против 13,33 %, в контроле — 4,76 %), арахнодактилия и склонность к кровотечениям отмечались только у детей I группы (23,33 и 6,67 % соответственно, в контроле не отмечалось). Определение уровня оксипролина в моче у детей с рецидивирующим пиелонефритом свидетельствует об усилении распада и выведения продуктов обмена коллагена в детском организме у 97 % обследованных, что значительно превышает частоту выделения оксипролина с мочой у детей с острым течением пиелонефрита (10 %) и указывает на выраженное нарушение катаболизма коллагена у детей, склонных к рецидивам пиелонефрита. Установлена достоверно высокая частота «дикого» генотипа АА — rs605143 гена коллагена COL4A1 у детей с рецидивирующим течением пиелонефрита по сравнению с данными лиц общепопуляционной контрольной группы (21,4 % против 4,8 %, p < 0,05). Наличие у ребенка генотипа АА полиморфного локуса rs605143 гена COL4A1 в пять раз увеличивает риск развития рецидивирования хронического пиелонефрита (отношение шансов 5,105, 95% доверительный интервал 0,12–0,87). Выводы. С целью прогнозирования у детей генетически детерминированной склонности к рецидивированию пиелонефрита рекомендуется проведение молекулярно-генетического тестирования генотипов AA и AG полиморфного локуса rs605143 гена коллагена COL4A1.
Background. In recent years, the clinical course of pyelonephritis in children is characterized by an increase in the number of latent and subclinical forms of the disease, increased resistance to traditional antibiotic therapy, which leads to chronic and recurrent pathological process in the kidneys. At the same time, the structure of nephropathy in children is characterized mainly by congenital and hereditary diseases, as well as diseases associated with hereditary predisposition, with a latent onset and torpid course. Particular attention should be paid to the children with manifestations of connective tissue dysplasia. The objective of the study: the search for possible clinical and laboratory markers associated with fibrillogenic disorders in children with different variants of pyelonephritis, and finding out the role of fibrillogenic disorders in the severity of pyelonephritis in children. Materials and methods. Sixty children with pyelonephritis aged 3 to 18 years were examined. According to the results of catamnestic observation, they were divided into two groups: І — 30 children with pyelonephritis, who had 3 or more episodes of recurrent pyelonephritis during the year, and ІІ — 30 children with pyelonephritis, who had no relapses during the year. The control group consisted of 42 somatically healthy children of the same age. All children underwent a routine comprehensive clinical and laboratory examination and determination of clinical and laboratory markers of fibrillogenic disorders (phenotypic signs of undifferentiated connective tissue dysplasia, hydroxyproline in urine). A molecular genetic testing of the AA and AG loci of the RS 605143 polymorphic locus of the COL4A1 gene was conducted. Results. In children with recurrent pyelonephritis, phenotypic signs of undifferentiated connective tissue dysplasia were more likely to be observed compared to those in children, who had only one episode of pyelonephritis during the year, namely: joint hypermobility (in 53 % of children versus 6.67 % in the comparison group, controls — 4.76 %), asthenic body structure (56.67 vs. 26.67 %, controls — 16.67 %), visual impairment (86.67 vs. 36.67 %, controls — 7.14 %), deformity of the chest (43.33 vs. 10 %, controls — 4.76 %), scoliostic posture (53.33 vs. 13.33 %, controls — 4.76 %), arachnodactylia and predisposition to bleeding were recorded only in children from group I (23.33 and 6.67 %, respectively, no cases in the control group). Determination of hydroxyproline level in the urine in children with recurrent pyelonephritis indicates an increased decay and excretion of collagen metabolism products in the child’s body in 97 % of the subjects, which significantly exceeds the rate of hydroxyproline urinary excretion in children with acute pyelonephritis (10 %), and indicates the significant disturbances of collagen catabolism in children prone to recurrent pyelonephritis. The probable higher frequency of the “wild” AA genotype — rs605143 gene of the collagen COL4A1 in children with recurrent pyelonephritis was found to be higher than that of the general population (21.4 vs. 4.8 %, p < 0.05). The presence of AA genotype of the rs605143 gene polymorphic locus of COL4A1 increases the risk of recurrence of chronic pyelonephritis by five times (odds ratio 5.105, 95% confidence interval 0.12–0.87). Conclusions. In order to predict a genetically determined predisposition to recurrent pyelonephritis in children, it is recommended to carry out molecular genetic testing for AA and AG genotypes of the polymorphic locus rs605143 gene of the COL4A1 collagen.
діти; дисплазія сполучної тканини; порушення фібрилогенезу; клініко-лабораторні маркери; оксипролін; колаген; пієлонефрит
дети; дисплазия соединительной ткани; нарушение фибриллогенеза; клинико-лабораторные маркеры; оксипролин; коллаген; пиелонефрит
children; connective tissue dysplasia; fibrillogenic disorders; clinical and laboratory markers; hydroxyproline; collagen; pyelonephritis
Вступ
Матеріали та методи
Результати та обговорення
/41-1.jpg)
/42-1.jpg)
/43-1.jpg)