Белки сурфактанта составляют 10,6 % от основной массы и представляют собой смесь сывороточных и несывороточных белков. Несывороточные белки принято называть SP-A, SP-B, SP-C и SP-D [30].
SP-A — гликопротеин, принадлежащий к семейству лектинов С-типа, весит 26–35 кДа и синтезируется на длинном плече 10-й хромосомы из двух генов: SFTPA1 и SFTPA2. Хотя ткань легкого является основным местом синтеза SP-A, белок обнаружен в кишечнике, эндокринной системе и среднем ухе. SP-D (43 кДа) также синтезируется на длинном плече 10-й хромосомы геном SFTPD, а общая структура SP-D аналогична общей структуре SP-A [15]. SP-D сопряжен с фосфатидилинозитолом и глюкозилцерамидом сурфактанта [21], имеет домен распознавания углеводов, способствуя агглютинации бактерий, вирусов и грибов [14].
Гидрофильные белки SP-A и SP-D хорошо экспрессируются в пре- и постнатальном периодах. SP-A играет важную роль в иммунной защите, фагоцитозе и киллинге бактериальных, вирусных и грибковых патогенов, практически не влияя на эластические свойства сурфактанта [26].
Другая группа белков, SP-B и SP-C, обусловливают эластические свойства сурфактанта. SP-B — гидрофобный полипептид, состоящий из 79 аминокислот, синтезируется геном SFTPB на 2-й хромосоме (2p12-p11.2). SP-B связывается с фосфолипидными бислоями и обладает как мембранными, так и фузогенными свойствами, что обеспечивает организацию фосфолипидных мембран в ламеллярных гранулах [22].
SP-C — самый гидрофобный белок сурфактанта, состоит из 35 аминокислот и синтезируется геном SFTPC на 8-й хромосоме. Высока вероятность связи SP-C с жидким бислоем дипальмитоилфосфатидилхолина. Кроме того, SP-C может вклиниваться в двухслойный фосфолипид, поддерживая монослой поверхностно-активного вещества в альвеоле, усиливать поглощение и катаболизм фосфолипидов альвеолоцитами II типа [3, 4, 30].
SP-B и SP-C синтезируются в клетках альвеолярного типа II в виде больших белков-предшественников (proSP-B и proSP-C) в эндоплазматическом ретикулуме, которые расщепляются протеолитическими ферментами с получением меньших, чрезвычайно гидрофобных пептидов (рис. 1). Затем они транспортируются через аппарат Гольджи в лизосомы, с последующим протеолизом белков-предшественников (proSP-B и proSP-C). Лизосома сливается с ламеллярной гранулой, зрелые белки встраиваются в поверхностно-активные фосфолипидные мембраны, и происходит секреция путем слияния мембраны ламеллярной гранулы с плазматической мембраной эпителиальной клетки, в результате чего содержимое выливается в альвеолярное пространство [26]. Богатое фосфолипидом содержимое собирается в трубчатый миелин, который служит резервуаром поверхностно-активного вещества во время дыхания и усиливает выведение липидов в альвеолярное пространство [30].
/177-1.jpg)
Во время дыхания пленка сурфактанта находится под высоким давлением при низком остаточном объеме легких, что способствует десорбции липидов и протеинов (SP-B, SP-C). Часть белков и жиров рециркулируется клетками II типа, где происходит эндоцитоз через лизосомы и транспортировка в ламеллярные гранулы, иные внеклеточно собираются в трубчатый миелин [25], в то время как остальные поглощаются макрофагами. Рециркуляция веществ сурфактанта объясняет длительный эффект заместительной терапии сурфактантом у преждевременно рожденных детей.
Еще один интегральный мембранный белок — ABCA3, состоящий из 1704 аминокислот, локализованный на внешней мембране ламеллярной гранулы, является членом большого семейства АТФ-связывающих кассетных белков, которые активно транспортируют различные вещества через биологические мембраны, включая липиды [9, 15]. ABCA3 функционирует для импорта фосфатидилхолина и фосфатидилглицерина из цитозоля в ламеллярное тело.
Таким образом, именно SP-B и SP-C играют наиболее существенную роль в эластической способности и поддержании остаточного объема легких и обусловливают формирование нормальных бислоев сурфактанта, поддерживая его поверхностно-восстановительные вещества и облегчая адсорбцию сурфактанта. ABCA3 также синтезируется и гликозилируется в эндоплазматическом ретикулуме, а затем транспортируется через аппарат Гольджи в лизосому на наружную мембрану ламеллярной гранулы [30].
Остановимся на генетической и гистологической характеристиках наследственного дефицита протеинов сурфактанта и клинической реализации у детей.
Наследственный дефицит SP-B впервые был описан в 1993 году у доношенного новорожденного с интерстициальным заболеванием легких (ИЗЛ) [31]. Ребенок умер в возрасте 5 месяцев от прогрессирующей дыхательной недостаточности, которая развилась вскоре после рождения и не разрешалась, несмотря на проведение механической вентиляции легких, введение сурфактанта и кортикостероидов, а также экстракорпоральную мембранную оксигенацию. Био–псия легких показала изменения, характерные для врожденного альвеолярного протеиноза. В семейном анамнезе имел место летальный исход у предыдущего ребенка в результате респираторного дистресс-синдрома (РДС) новорожденных. При генетическом анализе доказано, что мутация 121ins2 SFTPB в 4-м экзоне преждевременно активировала стоп-кодон в 6-м экзоне и препятствовала экспрессии зрелого SFTPB, что приводило к полному отсутствию РНК-носителя и белка SP-B в легком ребенка.
На сегодняшний день выявлено более 40 различных мутаций в гене SP-B [2], из них 2/3 — в 4-м экзоне. Родители и сиблинги детей с мутацией гена SFTPB клинических проявлений заболевания не имеют, а у ребенка заболевание возникает при мутации обеих аллелей, приводящей к полному отсутствию или потере функции SP-B.
Дефицит SP-B наследуется по аутосомно-рецессивному типу. Заболеваемость в Соединенных Штатах Америки составляет 1 : 1 000 000 новорожденных, из них частота мутации 121ins2 в 4-м экзоне — приблизительно 1 : 1000–3000 человек [32]. К сожалению, в мире диагностика наследственного дефицита SP-B проводится редко, а в некоторых странах вовсе нет доступа к анализу мутаций генов сурфактанта, что выражается в недостоверных эпидемиологических показателях.
Заболевание манифестирует с респираторного дистресс-синдрома у доношенных новорожденных. В бронхоальвеолярной лаважной жидкости (БАЛ) отмечается повышение фосфатидилинозитола и снижение фосфатидилглицерола, что обусловливает неэффективное обеспечение поверхностного натяжения альвеол [32]. Хотя временное улучшение может наблюдаться при введении экзогенного сурфактанта, трансплантация легких в настоящее время является единственным эффективным лечением [32]. Без трансплантации дети погибают в возрасте 3–6 месяцев [21].
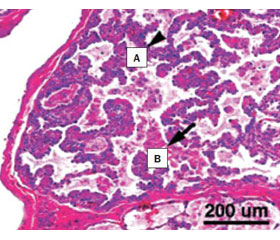
Гистологически дефицит SP-B манифестирует с врожденного альвеолярного протеиноза (за счет нарушения передачи сигналов GM-CSF в макрофаге, что препятствует рециркуляции сурфактанта) (рис. 2) и, реже, с инфантильного десквамативного интерстициального пневмонита (рис. 3).
/178-1.jpg)
Патоморфология сходна с приобретенным легочным альвеолярным протеинозом у взрослых (обу–словленным продукцией антител против гранулоцитарного макрофагального колониестимулирующего фактора (granulocyte-macrophage colony-stimulating factor, GM-CSF)) или легочным альвеолярным протеинозом у детей (инициированным мутацией в общей бета- или альфа-субъединице рецептора GM-CSF). Вместе с тем легочный альвеолярный протеиноз при наследственном дефиците SP-B проявляется накоплением в альвеолах гранулярного, эозинофильного вещества, липопротеинов, десквамированных альвеолоцитов II типа и пенистых альвеолярных макрофагов с нарушенной функцией. Однако отличительной особенностью дефицита SP-B является небольшое количество материала в альвеолах, вплоть до его полного отсутствия. При мутации в общей бета- или альфа-субъединице рецептора GM-CSF (легочным альвеолярным протеинозом у детей) альвеолярная архитектура сохраняется, и напротив, для морфологии мутации в гене SFTPB характерны утолщение альвеолярных перегородок и пролиферация фибробластов с воспалительными клеточными инфильтратами. Кроме того, у детей с наследственным дефицитом SP-В мальформированы ламеллярные гранулы и трубчатый миелин в виде дезорганизованных мультивезикулярных структур в клетках альвеолярного II типа [2, 32].
Удивительно, но у детей с недостаточностью SP-В в альвеолах накапливается частично обработанный proSP-C, что доказывает роль SP-B в обработке и транспорте proSP-C. Вместе с тем как proSP-B, так и зрелые SP-B у пациентов отсутствуют. В альвеолярном просвете также накапливается SP-A, и этому пока нет объяснения [32].
Дети с отсутствием накопления материала в альвеолах гистологически проявляют десквамативный интерстициальный пневмонит (рис. 4).
Наследственный дефицит SP-С впервые описан в 2001 году также у доношенного новорожденного с респираторным дистресс-синдромом [33]. У ребенка в возрасте 6 недель развились респираторные расстройства, которые включали тахипноэ и цианоз. По данным рентгенографии органов грудной клетки была выявлена гиперинфляция. Патологоанатомически был установлен диагноз неспецифического интерстициального пневмонита с хорошо сохранившимися утолщенными альвеолярными перегородками, гиперплазией альвеолярных клеток II типа, инфильтрацией интерстиция, состоящей из зрелых лимфоцитов и мио–фибробластов. Альвеолы были частично заполнены десквамированными клетками и альвеолярными макрофагами. Мать ребенка с раннего возраста страдала десквамативным интерстициальным пневмонитом, а дедушка по материнской линии умер от хронической болезни легких неизвестной этиологии.
Матери ребенка также проведена биопсия легкого, в ткани выявлены области диффузного фиброза и формирование сотового легкого, с участками лимфоцитарной и макрофагальной инфильтрации интерстиция.
Генетическое исследование доказало гетерозиготную мутацию гена SFTPC у ребенка, включающую замещение аденина гуанином в 4-м интроне, что привело к делеции гена, отвечающего за синтез 37 аминокислот. Аналогичная мутация выявлена и у матери [33]. РroSP-C присутствовал в небольших количествах в ткани легкого как у пациента, так и у его матери, а зрелый SP-C отсутствовал. Количество SP-B было нормальным.
В настоящее время идентифицировано более 40 различных мутаций в гене SFTPC [3]. Мутации включают миссенс-мутации в 3, 4 и 5-м экзонах proSP-C, которые приводят к нарушению переработки белка-предшественника (proSP-C) в зрелый пептид. В отличие от генетического дефицита SP-B интерстициальное заболевание легких, вызванное мутациями SFTPC, имеет аутосомно-доминантный тип наследования с переменной пенетрантностью (45 %) или мутацией de novo на одной аллели как спорадическое заболевание (55 %). Мутация треонина в кодоне 73 (I73T) в гене SFTPC составляет значительную часть мутаций SFTPC, ассоциированных с интерстициальной болезнью легких [4].
Частота вариантов мутации SFTPC в популяции в настоящее время неизвестна, что связано с различным возрастом манифестации и клиникой заболевания. Вместе с тем наследственный дефицит Sp-С чаще дебютирует в детском возрасте и крайне редко у взрослых.
Клиническая картина заболевания может быть различной [12]. Приблизительно от 10 до 15 % пациентов с мутацией SFTPC развивают респираторные симптомы в течение 1-го месяца жизни и 40 % — в возрасте от 1 до 6 месяцев жизни, причем средний возраст начала заболевания — от 2 до 3 месяцев. У новорожденных заболевание дебютирует с респираторного дистресс-синдрома [11], у детей до 2 лет — с симптомов диффузного заболевания легких, включая тахипноэ, ретракцию, гипоксемию и стагнацию физического развития [15]. Дети склонны к частым респираторным заболеваниям. У подростков с гетерозиготной мутацией в 5-м экзоне SP-C возможна манифестация ИЗЛ с клиники неспецифической интерстициальной пневмонии или обычного интерстициального пневмонита [15], а триггером может быть вирусная инфекция (например, респираторно-синцитиальный вирус, грипп A и B).
Вместе с тем прогрессирование заболевания индивидуально. Некоторым пациентам требуется трансплантация легкого в первые 2 года заболевания, в то время как другие имеют хорошую продолжительность жизни на фоне кислородозависимости или вовсе уменьшают потребность в кислороде, что свидетельствует о влиянии дополнительных генетических и экзогенных факторов, влияющих на начало и прогрессирование заболевания.
Гистологическая картина наследственного дефицита SP-С проявляется легочным альвеолярным протеинозом (рис. 5), неспецифическим (рис. 4) или десквамативным интерстициальным пневмонитом, хроническим пневмонитом новорожденных [32].
Так же, как и при наследственном дефиците –SP-В, легочный альвеолярный протеиноз выражается в диффузном повреждении альвеол различной степени тяжести, в гиперплазии альвеолярного эпителия и интерстиция с небольшой инфильтрацией лимфоцитами, пенистыми альвеолярными макрофагами с отложением гранулоцитов, холестерина в альвеолах [32].
У подростков с мутациями SFTPC и ИЗЛ наиболее распространенным гистопатологическим диагнозом является легочный фиброз.
Ламеллярные тела и трубчатый миелин могут быть нормальными (преимущественно у детей с мутацией в 4-м экзоне) или мальформированными, а SP-A, proSP-B, SP-B и SP-D, proSP-C присутствуют в достаточном количестве. Характерно накопление в клетке токсичного proSP-C, что способствует травматизации, стрессу эндоплазматического ретикулума и апоптозу. Гистологически это проявляется хроническим воспалением интерстиция и пневмофиброзом. У пациентов с мутациями 4-го экзона и 91-93del9 также выявляется снижение или отсутствие SP-C. И наоборот, при мутации E66K и I73T секретируются как зрелые SP-C, так и proSP-C, однако нарушается состав и функция фосфолипидов [32].
Наследственный дефицит ABCA3 впервые описан в 2004 году также у доношенного ребенка, тяжелый респираторный дистресс-синдром у которого привел к летальному исходу. Семейный анамнез демонстрировал заболевания легких у родственников. Гистологически выявлялась десквамативная интерстициальная пневмония.
Доказано, что дефицит ABCA3 наследуется аутосомно-рецессивно. Выявлено 150 различных мутаций 30 кодирующих экзонов в гене ABCA3, расположенном на 16-й (16p13.3) хромосоме [5]. В 2016 году открыта новая мутация ABCA3, R288K, увеличивающая риск развития ИЗЛ у детей [24]. В зависимости от местоположения мутации в гене патогенез заболевания различен и включает снижение экспрессии, недостаточный транспорт в ламеллярные гранулы, а также аномальное формирование и функцию фосфолипидной мембраны. В сурфактанте количество фосфатидилхолина и фосфатидилглицерола уменьшено, что нарушает поверхностное натяжение альвеол.
Недавно открыты миссенс-мутации в гене ABCA3, которые влияют на гомеостаз сурфактанта, нарушая локализацию внутриклеточного белка ABCA3 (c. 643C > A, p. Q215K, c. 2279T > G, p. M760R) и липидного переноса ABCA3 (c. 875A > T, p. E292V, c. 4164G > C, p. K1388N). Кроме этого, определены мутации, предрасполагающие к развитию интерстициальных заболеваний легких, несмотря на правильную локализацию и нормальный липидный перенос ABCA3 (c. 622C > T, p. R208W; c. 863G > A, p. R288K; c. 2891G > A, p. G964D) [1].
Пока единственная мутация, а именно замещение валина в кодоне 292 (E292V), расположенном в первой цитозольной петле белка ABCA3, идентифицирована у детей подросткового возраста и имеет относительно благоприятный прогноз. У этих пациентов заболевание дебютирует в грудном возрасте с формированием ИЗЛ и гистологией легочного альвеолярного протеиноза или десквамативной интерстициальной пневмонии. Дети выживают без трансплантации легких [22, 23]. В последнее время частота мутации E292V в 3–5 раз выше, чем мутаций в SFTPB (121ins2) или SFTPC (I73T) (< 0,4 %), а мутация E292V может быть негативным модифицирующим фактором респираторной патологии у взрослых и РДС у новорожденных.
Большинство случаев мутации ABCA3 манифестирует с РДС в неонатальном периоде или в грудном возрасте. Средний возраст дебюта заболевания составляет 1,3 ± 0,5 месяца [7, 8], с летальным исходом без трансплантации легких [22].
Гистологическая картина наследственного дефицита ABCA3 отображает наследственный альвеолярный протеиноз, неспецифическую или десквамативную интерстициальную пневмонию в периоде новорожденности [9]. Отличительной особенностью является то, что в альвеолоцитах II типа ламеллярные гранулы мелкие, с плотно депонированными фосфолипидами. Белки сурфактанта SP-A, SP-B, proSP-B, proSP-C и SP-D в достаточном количестве. Однако у части пациентов зрелого SP-B недостаточно, что акцентирует роль ABCA3 в трансформации proSP-B в SP-B. В отличие от дефицита SP-B proSP-C присутствует в цитоплазме альвеолоцитов II типа и не встречается в альвеолах.
В заключение описания клинико-морфологической картины отметим, что мутации в гене ABCA3 вызывают существенную дисфункцию сурфактанта в результате нарушения образования SP-B и SP-C, а также депонирования и секреции фосфолипидов. Наиболее неблагоприятными считаются мутации SP-B и ABCA3 (кроме E292V), что обычно проявляется респираторным дистресс-синдромом в неонатальном периоде. Летальный исход при данной патологии наступает в первые месяцы жизни, а механическая вентиляция легких, введение сурфактанта и даже экстракорпоральная мембранная оксигенация неэффективны. Гистологически данные нозологические формы чаще манифестируют в виде легочного альвеолярного протеиноза, неспецифической или десквамативной интерстициальной пневмонии. Однако отличительной особенностью наследственного дефицита протеинов SP-B и ABCA3 сурфактанта считается относительно небольшое накопление клеток, белков-предшественников (при дефиците SP-B) и продуктов распада в альвеолах и утолщение межальвеолярной перегородки, а также мальформированные ламеллярные гранулы и трубчатый миелин.
Сходная гистологическая картина SFTPB и ABCA3 обусловливает необходимость их дифференциальной диагностики. Недостаточность SP-B нарушает образование хорошо организованных концентрических колец фосфолипидных мембран, которые обычно наблюдаются в небольшой ламеллярной грануле. Вместо этого в цитозоле альвеолоцитов II типа, а также в альвеолярном просвете отмечаются большие неорганизованные мультивезикулярные тела. Напротив, небольшие мальформированные ламеллярные тельца с плотно сформированными мембранами фосфолипидов и включениями наблюдаются в альвеолоцитах II типа у пациентов с мутациями ABCA3. Пока не описаны ультраструктурные аномалии, характерные для мутации SFTPC, хотя сообщалось о неправильных или дезорганизованных ламеллярных гранулах.
Следует помнить, что аномальные пластинчатые тела также обнаруживаются при некоторых заболеваниях, таких как синдромы Чедиака — Хигаси и Германски — Пудлака. Однако для этих заболеваний характерен фенотип гигантских ламеллярных гранул, что отличает их от мутации SFTPB и ABCA3.
Мутации SP-С имеют более благоприятное течение и прогноз. Возможно развитие респираторного дистресс-синдрома в неонатальном периоде, но чаще болезнь дебютирует с интерстициального заболевания легких в раннем и дошкольном возрасте на фоне вирусной инфекции. Описаны случаи манифестации в подростковом возрасте. Отличительной особенностью клиники дефицита является зависимость от возраста дебюта заболевания. Клинически дефицит проявляется ИЗЛ, а гистологическая картина сходна с недостаточностью SP-B и ABCA3, однако с относительно замедленным (месяцы, иногда — годы) поражением межальвеолярных перегородок и накоплением продуктов сурфактанта и клеток. Отличительной ультраструктурной чертой мутации SP-С считаются преимущественно нормальные ламеллярные тела и трубчатый миелин, а также накопление токсичного proSP-C в клетке, что способствует травматизации, стрессу эндоплазматического ретикулума и апоптозу.
Диагностика мутаций протеинов сурфактанта включает [13, 16]:
1. Анализ анамнеза.
2. Генетическую диагностику.
3. Бронхоскопию с бронхоальвеолярным лаважем.
4. Компьютерную томографию высокого разрешения.
5. Биопсию с иммуногистохимическим и ультраструктурным исследованием (электронная микроскопия) легочной ткани.
Среди особенностей анамнеза дефицита белков сурфактанта имеет место семейная история респираторного дистресс-синдрома, интерстициального и хронического заболевания легких.
Генетическая диагностика (а именно — исследование мутаций в генах SFTPB, ABCA3) проводится всем доношенным новорожденным, которые развили респираторный дистресс-синдром (сильная рекомендация, высокий уровень доказательности). И при условии формирования ИЗЛ у ребенка до 2 лет рекомендовано исследование мутаций генов SFTPC и ABCA3 (сильная рекомендация, высокий уровень доказательности). Для младенцев с альвеолярным протеинозом, имеющих отрицательный результат исследования мутаций SFTPC и ABCA3, рекомендовано дополнительно проводить генетическое тестирование CSF2RA и CSF2RB (слабая рекомендация, низкий уровень доказательности). А при положительных тестах на CSF2RA и CSF2RB возможно определение гранулоцитарно-макрофагального колониестимулирующего фактора (GM-CSF) (слабая рекомендация, низкий уровень доказательности). Все генетические исследования необходимо проводить в сертифицированных лабораториях. К сожалению, в настоящее время исследование дефицита белков сурфактанта доступно в нескольких лабораториях США и Европы, а генетические исследования CSF2RA и CSF2RB проводятся только в контексте исследований [25].
Бронхоскопия с бронхоальвеолярным лаважем не может заменить биопсию легочной ткани. Однако иногда используется для дифференциальной диагностики ИЗЛ (слабая рекомендация, низкий уровень доказательности).
Биопсия с иммуногистохимическим и ультраструктурным исследованием (электронная микроскопия) легочной ткани — золотой стандарт диагностики патофизиологии респираторного нарушения. Использование иммуногистохимического исследования с помощью моноспецифических антител позволяет дифференцировать частично обработанные и/или неправильные формы пропептидов. Иммуногистохимический анализ особенно полезен при определении диагноза генетического дефицита SP-B, поскольку для мутаций SFTPB описаны отдельные иммуногистохимические структуры для зрелой экспрессии SP-B и proSP-C. В настоящее время иммуногистохимические исследования пока недоступны в Украине.
К сожалению, до настоящего времени протокол лечения наследственного дефицита протеинов сурфактанта не разработан. Представляем алгоритм менеджмента, основанный на современных литературных источниках (рис. 6) [17–22].
Таким образом, протеины SP-B, SP-C и ABCA3 способствуют формированию нормальных бислоев сурфактанта, поддержанию эластической способности и остаточного объема легких. Наиболее неблагоприятные мутации SP-B и ABCA3 обычно проявляются респираторным дистресс-синдромом в неонатальном периоде с высокой вероятностью летального исхода. Мутации SP-С имеют торпидное течение и прогноз, а заболевание дебютирует с интерстициального заболевания легких в раннем и дошкольном возрасте на фоне вирусной инфекции. Диагностика наследственного дефицита протеинов сурфактанта ограничена генетическим исследованием, биопсией с иммуногистохимической и ультраструктурной оценкой, что может обеспечить правильную тактику лечения пациентов.
Конфликт интересов. Авторы заявляют об отсутствии какого-либо конфликта интересов при подготовке данной статьи.
Список литературы
1. Schindlbeck U. ABCA3 missense mutations causing surfactant dysfunction disorders have distinct cellular phenotypes / U. Schindlbeck, T. Wittmann, S. Höppne // Hum. Mutat. — 2018. — № 39(6). — P. 841-850.
2. Delayed Presentation and Prolonged Survival of a Child with Surfactant Protein B Deficiency / J.A. López-Andreu, A.D. Hidalgo-Santos, M.A. Fuentes-Castelló // J. Pediatr. — 2017. — № 190. — P. 268.
3. Brasch F. Interstitial lung disease in a baby with a de novo mutation in the SFTPC gene / F. Brasch, M. Griese, M. Tredano et al. // Eur. Respir. J. — 2004. — № 24. — P. 30.
4. Hawkins A. A non-BRICHOS SFTPC mutant (SP-CI73T) linked to interstitial lung disease promotes a late block in macroautophagy disrupting cellular proteostasis and mitophagy / A. Hawkins, S.H. Guttentag, R. Deterding et al. // Am. J. Physiol. Lung. Cell. Mol. Physiol. — 2015. — № 308. — P. 33.
5. Kröner C. Lung disease caused by ABCA3 mutations / C. Kröner, T. Wittmann, S. Reu et al. // Thorax. — 2017. — № 72. — P. 213.
6. Wambach J.A. Genotype-phenotype correlations for infants and children with ABCA3 deficiency / J.A. Wambach, A.M. Casey, M.P. Fishman et al. // Am. J. Respir. Crit. Care Med. — 2014. — № 189. — P. 1538.
7. Thavagnanam S. Variable clinical outcome of ABCA3 deficiency in two siblings / S. Thavagnanam, E. Cutz, D. Manson et al. // Pediatr. Pulmonol. — 2013. — № 48. — P. 1035.
8. Hallik M. Different course of lung disease in two siblings with novel ABCA3 mutations / M. Hallik, T. Annilo, M.L. Ilmoja // Eur. J. Pe–diatr. — 2014. — № 173. — P. 1553.
9. Wambach J.A. Single ABCA3 mutations increase risk for neonatal respiratory distress syndrome / J. A.Wambach, D.J. Wegner, K. Depass et al. // Pediatrics. — 2012. — № 130. — P. 1575.
10. Litao M.K. A novel surfactant protein C gene mutation associated with progressive respiratory failure in infancy / M.K. Litao, D. Jr. Hayes, S. Chiwane et al. // Pediatr. Pulmonol. — 2017. — № 52. — P. 57.
11. Avital A. Natural history of five children with surfactant protein C mutations and interstitial lung disease / A. Avital, A. Hevroni, S. Godfrey et al. // Pediatr. Pulmonol. — 2014. — № 49. — P. 1097.
12. Kröner C. Genotype alone does not predict the clinical course of SFTPC deficiency in paediatric patients / C. Kröner, S. Reu, V. Teusch et al. // Eur. Respir. J. — 2015. — № 46. — P. 197.
13. Kurland G. An official American Thoracic Society clinical practice guideline: classification, evaluation, and management of childhood interstitial lung disease in infancy / G. Kurland, R.R. Deterding, J.S. Hagood et al. // Am. J. Respir. Crit. Care Med. — 2013. — № 188. — P. 376.
14. Gower W.A. Candidate gene analysis of the surfactant protein D gene in pediatric diffuse lung disease / W.A. Gower, L.M. Nogee // J. Pediatr. — 2013. — № 163. — P. 1778.
15. Henderson L.B. Large ABCA3 and SFTPC deletions resulting in lung disease / L.B. Henderson, K. Melton, S. Wer et al. // Ann. Am. Thorac. Soc. — 2013. — № 10. — P. 602.
16. Hevroni A. Infant pulmonary function testing in chronic pneumonitis of infancy due to surfactant protein C mutation / A. Hevroni, A. Goldman, C. Springer // Pediatr. Pulmonol. — 2015. — № 50. — P. 17.
17. Hepping N. Successful treatment of neonatal respiratory fai–lure caused by a novel surfactant protein C p.Cys121Gly mutation with hydroxychloroquine / N. Hepping, M. Griese, P. Lohse et al. // J. Perinatol. — 2013. — № 33. — P. 492.
18. Tan J.K. ABCA3 lung disease in an ex 27 week preterm infant responsive to systemic glucocorticosteroids / J.K. Tan, C. Murray, –A. –Schultz // Pediatr. Pulmonol. — 2016. — № 51. — P. 1.
19. Williamson M. Ten-year follow up of hydroxychloroquine treatment for ABCA3 deficiency / M. Williamson, C. Wallis // Pediatr. Pulmo–nol. — 2014. — № 49. — P. 299.
20. Thouvenin G. Diffuse parenchymal lung disease caused by surfactant deficiency: dramatic improvement by azithromycin / G. Thouvenin, N. Nathan, R. Epaud, A. Clement // BMJ Case Rep. — 2013. — № 3. — P. 1-3.
21. Eldridge W.B. Outcomes of Lung Transplantation for Infants and Children with Genetic Disorders of Surfactant Metabolism / W.B. Eldridge, Q. Zhang, A. Faro et al. // J. Pediatr. — 2017. — № 184. — P. 157.
22. Liptzin D.R. Chronic ventilation in infants with surfactant protein C mutations: an alternative to lung transplantation / D.R. Liptzin, T. Patel, R.R. Deterding // Am. J. Respir. Crit. Care Med. — 2015. — № 191. — P. 1338.
23. Naderi H.M. Single mutations in ABCA3 increase the risk for neonatal respiratory distress syndrome in late preterm infants (gestational age 34-36 weeks) / H.M. Naderi, J.C. Murray, J.M. Dagle // Am. J. Med. Genet. A. — 2014. — № 164. — P. 2676.
24. Wittmann T. Increased risk of interstitial lung disease in children with a single R288K variant of ABCA3 / T. Wittmann, S. Frixel, S. Höppner et al. // Mol. Med. — 2016. — № 22. — P. 183-191.
25. Tanaka T. Adult-onset hereditary pulmonary alveolar proteinosis caused by a single-base deletion in CSF2RB / T. Tanaka, N. Motoi, Y. Tsuchihashi et al. // J. Med. Genet. — 2011. — № 48. — P. 250.
26. van Moorsel C.H. SFTPA2 Mutations in Familial and Sporadic Idiopathic Interstitial Pneumonia / C.H. van Moorsel, L. Ten Klooster, M.F. van Oosterhout et al. // Am. J. Respir. Crit. Care Med. — 2015. — № 192. — P. 1249.
27. Nathan N. Germline SFTPA1 mutation in familial idiopa–thic interstitial pneumonia and lung cancer / N. Nathan, V. Giraud, C. Picard // Hum. Mol. Genet. — 2016. — № 25. — P. 1457.
28. Silveyra P. Genetic variant associations of human SP-A and SP-D with acute and chronic lung injury / P. Silveyra, J. Floros // Front. Biosci. (Landmark Ed). — 2012. — № 17. — P. 407.
29. Somaschini M. Genetic surfactant dysfunction in newborn infants and children with acute and chronic lung disease / M. Somaschini, S. Presi, M. Ferrari et al. // Journal of Pediatric and Neonatal Individualized Medicine. — 2017. — № 6(1). — P. 1-8.
30. Chakraborty M. Pulmonary surfactant in newborn infants and children / M. Chakraborty, S. Kotecha // Breathe. — 2013. — № 9(6). — P. 477.
31. Nogee L.M. Brief report: deficiency of pulmonary surfactantprotein B in congenital alveolar proteinosis / L.M. Nogee, D.E. de Mello, L.P. Dehner et al. // N. Engl. J. Med. — 1993. — № 328. — P. 406-410.
32. Wert S.E. Genetic Disorders of Surfactant Dysfunction / S.E. Wert, J.A. Whitsett, L.M. Nogee // Pediatr. Dev. Pathol. — 2009. — № 12(4). — P. 253-274.
33. Nogee L.M. A mutation in the surfactantprotein C gene associated with familial interstitial lung disease / L.M. Nogee, A.E. Dunbar, S.E. Wert et al. // N. Engl. J. Med. — 2001. — № 344. — P. 573-579.

/178-2.jpg)
/180-1.jpg)