Глицин — простейшая аминокислота, не имеющая стереоизомеров, может выступать в качестве нейротрансмиттера в головном мозге, ингибируя деятельность ствола и спинного мозга, но возбуждая кору головного мозга. Глицин метаболизируется в митохондриях до конечных продуктов аммиака и двуокиси углерода через систему расщепления глицина (glycine cleavage system — GCS), ферментный комплекс, состоящий из четырех субъединиц (Р-белка, Н-белка, Т-белка, L-белка), дефекты которых могут вызывать некетотическую гиперглицинемию [1].
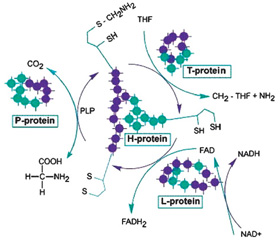
Глициновая энцефалопатия (некетотическая гиперглицинемия, nonketotic hyperglycinemia — NKH) — редкое генетическое метаболическое заболевание с аутосомно-рецессивным типом наследования, характеризуется дефицитом активности фермента расщепления глицина и, как следствие, накоплением большого количества глицина во всех тканях и жидкостях организма, включая головной мозг. В основе некетотической гиперглицинемии лежит мутация генов GLDC (9p24.1), кодирующий P-белок GCS (70–75 % случаев), AMT (3p21.31) кодирующий T-белок GCS (~ 20 % случаев) и GCSH (16q23.2) кодирующий H-белок GCS (< 1 % случаев) (рис. 1). Эти гены кодируют P-белковые и T-белковые компоненты ферментативной системы расщепления глицина. Пять процентов людей с глициновой энцефалопатией имеют мутации в других генах [2, 3].
Приводим собственное клиническое наблюдение девочки, 13 месяцев, госпитализированной в клинику с диагнозом «симптоматическая эпилепсия (частые полиморфные припадки), грубая задержка психомоторного развития, глазодвигательные нарушения». При поступлении мать предъявляла жалобы на усиление приступов эпилепсии на фоне приема антиконвульсантов (фармакорезистентность), утрату приобретенных навыков, грубую задержку психомоторного развития.
Из данных анамнеза выяснено, что ребенок от 1-й беременности, протекавшей на фоне острой респираторной инфекции, герпетической инфекции в 1-м триместре, дефицитной анемии во 2-м триместре. От первых срочных родов в сроке 41 неделя, осложненных тугим обвитием пуповиной вокруг шеи. Проводилась искусственная вентиляция легких в течение 2 суток по поводу внутриутробной пневмонии. К 3 месяцам девочка приобрела навыки удержания головы, поворота на бок, слежения за игрушкой. Однако на 4-м месяце появились и участились эпизоды судорожных подергиваний. Ребенок консультирован неврологом, инфекционистом выявлена цитомегаловирусная инфекция, в связи с чем проведено лечение внутривенным ацикловиром и иммуноглобулином. Получена непродолжительная положительная динамика в виде отсутствия судорожных приступов. С 8 месяцев эпилептические припадки возобновились, девочка продолжала отставать в психомоторном развитии, нарастала гипотония, утраченные навыки не приобретались. Согласно данным семейного анамнеза, троюродный брат пробанда по материнской линии умер в возрасте 12 лет на фоне прогрессирующего неврологического заболевания (с формированием детского церебрального паралича), троюродная сестра пробанда родилась с врожденными аномалиями кисти (трехпалость) и патологией органов зрения.
При поступлении в клинику отмечалось отставание в физическом развитии (масса тела — 7000 г, длина — 75 см). Девочка занимала вынужденное положение в постели с запрокинутой головой, отмечались миоклонические и тонические припадки до 25–30 серий. Ребенок самостоятельно не сидел, не переворачивался, не прослеживал предмет, не брал и не удерживал в руках игрушку. Вместе с тем в руках отмечались единичные хаотические движения, клонус стоп. При постановке на ножки ребенок не давал опору. Отсутствовало речевое развитие. Обращали на себя внимание множественные дизморфии: близко расположены глаза, гипертелоризм сосков, «кукольное лицо», узкая нижняя челюсть, пупочная грыжа, дизморфичные ушные раковины, низкий уровень роста волос, выступающее над уровнем кожи пятно кофейного цвета диаметром 0,5–0,7 см на передней поверхности брюшной стенки. От ребенка исходил резкий запах мочи. В соматическом статусе обращало на себя внимание небольшое увеличение печени: ее край пальпировался ниже реберной дуги на 2,5 см, гладкий, эластичной консистенции.
Проведены инструментальное и лабораторное обследования. На электрокардиографии выявлена синусовая тахикардия (частота сердечных сокращений — 136 ударов в 1 минуту), отмечалось нарушение процессов реполяризации.
Результаты эхоэнцефалографии демонстрировали нормотензию, М-эхо справа = М-эхо слева = 59, ширина М-эхо = 4,0, смещения не выявлено. По результатам электроэнцефалографии (ЭЭГ) определялись грубые нарушения паттерна ЭЭГ, выраженное снижение биоэлектрической активности (диффузное), снижение тормозных корковых влияний; отмечалась пароксизмальная активность в виде нечастых волн низкой амплитуды, признаки нижнестволовой дисфункции.
В биохимических показателях активность лактатдегидрогеназы была несколько повышена — 368,8 ЕД/л (норма — 12–295 ЕД/л).
Проведено одновременное исследование концентрации глицина в плазме крови и спинномозговой жидкости (СМЖ). Уровень глицина в плазме крови составлял 365 мкмоль/л, в спинномозговой жидкости — 70 мкмоль/л. Исследование концентрации органических кислот в моче позволило исключить кетотическую гиперглицинемию.
Результаты исследования глицина в крови дали основание предположить глициновую энцефалопатию, в связи с чем проведено молекулярно-генетическое тестирование GLDC, AMT, GCSH. Выявлена мутация GLDC (9p24.1).
На основании клиники, течения заболевания, лабораторных исследований и молекулярно-генетического тестирования ребенку установлен диагноз «глициновая энцефалопатия (инфантильная тяжелая форма). Симптоматическая эпилепсия, частые полиморфные припадки. Грубая задержка психомоторного развития».
Обсуждение
Частота некетотической гиперглицинемии составляет 1 : 76 000. Среди новорожденных в Финляндии NKH встречается с частотой 1 : 55 000 (1 : 12 000 — в Северной Финляндии), в Колумбии и Канаде — 1 : 63 000. Высокий уровень NKH регистрируется в арабских семьях и в Израиле [4].
Клинические проявления NKH. Большинство глициновых энцефалопатий развивается в неонатальном периоде (85 % — неонатальная тяжелая форма и 15 % — неонатальная аттенуированная (легкая) форма). Неонатальные формы начинают проявляться в первые часы и дни жизни в виде прогрессирующей вялости, гипотонии и миоклонических судорог, ведущих к апноэ и, часто, к смерти. Выжившие дети имеют глубокую умственную отсталость и трудно контролируемые судороги. В более старшем возрасте заболевание манифестирует с аттенуированного (легкого) течения (50 %), что проявляется гипотонией, задержкой развития и судорогами [6].
Как отмечалось выше, глициновая энцефалопатия подразделяется на две формы — неонатальную и инфантильную (манифестирует у детей до 2 лет), которые, в свою очередь, подразделяются на неонатальную тяжелую, неонатальную аттенуированную, инфантильную тяжелую, инфантильную аттенуированную и атипичную (редкую) формы.
Неонатальная (классическая) тяжелая форма отличается прогредиентным течением заболевания с первых часов жизни ребенка. Довольно быстро прогрессируют летаргия и гипотония, появляются миоклонические судороги, приводящие к апноэ и зачастую заканчивающиеся летальным исходом при отсутствии поддержания витальных функций. Дети, выжившие в неонатальном периоде, имеют грубые нарушения психомоторного развития.
Неонатальная аттенуированная форма у детей старше первого года жизни протекает значительно легче: дети имеют IQ > 60, возможны дефицит внимания и синдром гиперактивности. Редко отмечаются судороги, эпизоды летаргии, провоцируемые инфекционными агентами [6].
Для инфантильной формы не характерны эпизоды летаргии. Дебютирует заболевание гипотонией, с последующей задержкой психомоторного развития и инфантильными приступами от легкого до фармакорезистентного течения. У детей с тяжелым течением инфантильной формы к шестимесячному возрасту развиваются фармакорезистентные эпилептические припадки, бульбарные нарушения, прогрессирующие спастические параличи, приводящие к деформации скелета, патологической установке нижних конечностей, вынужденному положению в постели. Пациенты отстают в психомоторном и физическом развитии [2].
Аттенуированное течение инфантильной формы вариабельно. В клиническом симптомокомплексе возможны негрубые неврологические нарушения (эпиприпадки, синдром гипервозбудимости, хорееподобые движения) [2]. Несмотря на благоприятное течение заболевания, в анамнезе могут быть эпизоды тяжелой летаргии (что, вероятно, может провоцироваться приемом вальпроатов, поскольку они повышает концентрацию глицина в сыворотке). Некоторые из этих случаев были зарегистрированы как умеренная эпизодическая форма [6].
Клинические проявления атипичной формы глициновой энцефалопатии варьируются. Манифестация данной формы регистрируется в детском возрасте, редко — у взрослых. Характерными клиническими проявлениями заболевания являются спастический парапарез, атрофия зрительного нерва. У 50 % пациентов не выявляются судороги и когнитивные нарушения, у 28 % больных регистрируется умеренно выраженное снижение интеллекта с хореоатетозом, у 14,2 % — грубое снижение интеллекта и редкие эпиприпадки. В литературе представлены наблюдения двух новорожденных с легочной гипертензией, у которых развились пластический бронхит и прогрессирующее повреждение нейронов с формированием лейкодистрофии. Дети погибли в возрасте до 18 месяцев. У обоих детей изменения на электроэнцефалографии выявлены не были. Активность фермента расщепления глицина была нормальной (аутопсийный замороженный материал ткани печени), GLDC, AMT или GCSH мутаций генов не выявлено [9]. Позже у данных пациентов идентифицированы мутации генов NFU1 [10].
Ниже представлены клинические особенности течения глициновой энцефалопатии у детей с различными формами заболевания.
Диагностика. Ниже представлен алгоритм диагностики глициновой энцефалопатии, рекомендованный Национальным институтом здоровья США [1] (рис. 2).
Тестирование I уровня. Детям с подозрением на глициновую энцефалопатию проводят количественный аминокислотный анализ, который показывает концентрацию глицина в плазме и стерильной спинномозговой жидкости (количество лейкоцитов, эритроцитов и белка в СМЖ нормальное). Отмечено, что концентрация серина в СМЖ может быть снижена у детей с некетотической гиперглицинемией, при этом концентрация треонина не повышается. Изолированное повышение уровня глицина в спинномозговой жидкости и нарушение соотношения уровня глицина в плазме и СМЖ дают основание предположить глициновую энцефалопатию. Профиль аминокислот в моче у детей с глициновой энцефалопатией обычно нормальный. Интерпретация результатов, полученных на I уровне диагностики глициновой энцефалопатии, представлена в табл. 2.
/86-1.jpg)
/88-2.jpg)
Тестирование II уровня включает молекулярно-генетическое тестирование биаллельных генов: GLDC, AMT, GCSH (одновременно) посредством секвенирования и целевого анализа делеции, особенно GLDC гена. До 5 % людей с недостаточной ферментативной активностью расщепления глицина не имеют патогенного варианта в любом из трех генов. У этих пациентов исследуются патогенные варианты в генах, кодирующих пируватдегидрогеназный комплекс (липоат и пиридоксальфосфат), и транспорт глицина в астроцит (GLYT1), где расположен фермент GCS.
Тестирование III уровня проводится у пациентов с подозрением на глициновую энцефалопатию, не подтвержденным на I и II уровнях. Проводят клиновидную биопсию печени с получением не менее 80 мг материала. После измеряют активность GCS фермента (рис. 3). Фермент является лабильным, поэтому его уровень определяется быстро, в противном случае рекомендована глубокая заморозка.
Использование образцов периферической крови, культивируемых лимфобластами вируса Эпштейна — Барр, неэффективно [12].
Тестирование IV уровня может проводиться дополнительно и включает идентификацию недостаточности специфического протеина, С-глицина в выдыхаемом воздухе (перспективное направление), магнитно-резонансную спектроскопию (используется для неинвазивного измерения уровней глицина головного мозга).
Дифференциальная диагностика. Диагноз «транзиторная глициновая энцефалопатия» противоречивый. Некоторые исследователи описывают случаи транзиторной глициновой энцефалопатии у новорожденных с судорогами и снижением волновой активности на электроэнцефалограмме при нормальном или повышенном уровне глицина в плазме и спинномозговой жидкости, гетерозиготность по патогенным вариантам GLDC, GCSH генов или отсутствие мутаций указанных генов, с исходом в некетотическую гиперглицинемию, или ребенок развивался нормально. В настоящее время не выявлено достоверных прогностических признаков развития глициновой энцефалопатии у данной категории детей.
Гиперглицинемия может быть кетотической и является результатом лечения вальпроатами (могут обратимо увеличить концентрацию глицина в спинномозговой жидкости до ≥ 60 мкмоль/л у лиц, не имеющих глициновой энцефалопатии). В патогенезе обоих состояний лежит вторичное снижение активности фермента GCS печени, что лабораторно имитирует глициновую энцефалопатию.
Кетотическую гиперглицинемию можно наблюдать при пропионовой, метилмалоновой изовалериановой ацидемиях и дефиците β-кетотиолазы. Газовая хроматография/масс-спектрометрия позволяют установить уровень аминокислотных нарушений. Сообщалось о единственном пациенте с глицериновой ацидурией, сопровождающейся повышенными уровнями глицина, и недостаточной ферментативной активностью GCS.
Гиперглицинурия. Гиперглицинурию можно наблюдать при гиперпролинемии I или II типа, семейной иминоглицинурии и доброкачественной гиперглицинурии, что является результатом незрелости реабсорбции почечного глицина. Пациенты, гомозиготные по нулевой аллели, имеют высокую аффинность для пролина, гидроксипролина и глицина в проксимальном почечном канальце и клинически проявляют иминоглицинурию, тогда как гетерозиготы (носители) SLC36A2 и SLC6A18 имеют изолированную глицинурию (бессимптомную).
Недостаточность пиридоксинфосфатоксидазы. Лица с недостаточностью пиридоксинфосфатоксидазы имеют пиридоксаль-5-фосфатную (PLP) зависимую эпилепсию, летаргию, возможно, апноэ. Часто отмечается повышение глицина и треонина в сыворотке и спинномозговой жидкости (в плазме большее количество глицина, чем в СМЖ), а иногда и снижение метаболизма моноамина [13]. Концентрация пиридоксаль-5'-фосфата в СМЖ очень низкая.
Неонатальные судороги. Дифференциальная диагностика для неонатальных судорог широка и включает несколько генетических и метаболических состояний [12]: пероксосерные нарушения; дефицит сульфидоксидазы и кофактора молибдена (характеризуется очень низкой концентрацией цистеина при количественном определении аминокислот в крови и низкой концентрацией сывороточной мочевой кислоты); дефицит фосфоглицератдегидрогеназы, нарушение метаболизма серина; патогенные варианты в CDKL5; перинатальная гипоксически-ишемическая травма с повышением концентрации глицина в СМЖ и повышенным соотношением глицина СМЖ/плазма (отличием перинатальной гипоксически-ишемической травмы является повышение уровня белка и эритроцитов в СМЖ).
Мониторинг глициновой энцефалопатии включает магнитно-резонансную томографию (МРТ) головного мозга у новорожденных, проведение ЭЭГ (гипсаритмия), оценку психомоторного, физического, речевого развития, неврологического статуса. Диффузионно-взвешенные магнитно-резонансные изображения при МРТ головного мозга у детей с неонатальными формами демонстрируют симметричные высокосигнальные поражения, коррелирующие с вакуолизацией миелина. Характерна демиелинизация лимбических структур и ствола головного мозга [14]. Наличие пороков развития мозга является прогностическим неблагоприятным признаком и коррелирует со степенью тяжести заболевания. Часто выявляются пороки развития головного мозга, наиболее распространенные из которых — гипотрофия или агенезия мозолистого тела. Для инфантильной и атипичной форм более характерно торможение миелинизации и атрофия коры головного мозга.
Лечение. На сегодняшний день не существует эффективного лечения тяжелой глициновой энцефалопатии. Основная задача лечения заключается в снижении концентрации глицина бензоатом натрия в плазме путем блокирования глицинергических рецепторов на сайте рецептора N-метил-D-аспартата (NMDA). Бензоат натрия назначается перорально в дозе 250–750 мг/кг/сут в первые сутки. Дозу постепенно увеличивают на 50 мг/кг/сут до нормализации глицина в плазме (120–300 мкмоль/л). Бензоат натрия снижает концентрацию глицина плазмы до нормы, однако не нормализует концентрацию глицина спинномозговой жидкости. Пациентам с легкой формой заболевания назначают более низкие дозы бензоата натрия (200–450 мг/кг/сут). Применение бензоата натрия часто ассоциируется с гастритом, что может потребовать перорального введения антацидов, антагонистов Н2-рецепторов или ингибиторов протонной помпы. Антагонисты рецепторов N-метил-D-аспартата включают декстрометорфан или кетамин и фелбамат (противопоказаны при наличии в анамнезе судорог). Декстрометорфан (атуссин, гликодин, вокасепт) назначают в дозе 5–15 мг/кг/сут. Циметидин замедляет метаболизм декстрометорфана и не должен использоваться в комбинированном лечении. Кетамин вводится внутривенно в начальной дозе 0,7–2 мг/кг. Фелбамат (прозерин) рекомендован в дозе 0,05 мг (0,1 мл) на 1 год жизни, но не более 0,375 мг (0,75 мл) на одну инъекцию.
Противосудорожная терапия важна для улучшения качества жизни пациентов. При миоклонических приступах рекомендованы бензодиазепины. Стандартные противоэпилептические препараты, такие как фенобарбитал или фенитоин, эффективны при инфантильных формах и слабо эффективны в периоде новорожденности. Фелбамат успешно используется у некоторых детей с тяжелыми припадками.
Кетогенная диета снижает количество глицина, что должно учитываться при подборе дозы бензоата [14].
Для некоторых пациентов старшего возраста с тяжелой глициновой энцефалопатией и трудно контролируемыми судорогами применяется стимулятор блуждающего нерва.
У детей с бульбарным синдромом рекомендована установка гастростомы. При наличии гастроэзофагеального рефлюкса показана ниссенская фундопликация, что значительно снижает риск аспирационной пневмонии.
Выводы
Таким образом, глициновая энцефалопатия — редкое генетическое метаболическое заболевание с аутосомно-рецессивным типом наследования, характеризуется дефицитом активности фермента расщепления глицина и, как следствие, накоплением большого количества аминокислоты во всех тканях и жидкостях организма, включая головной мозг. Заболевание подразделяется на две формы — неонатальную и инфантильную (манифестирует у детей до 2 лет), которые, в свою очередь, подразделяются на неонатальную тяжелую, неонатальную аттенуированную, инфантильную тяжелую, инфантильную аттенуированную и атипичную (редкую) формы. Одновременное исследование глицина в плазме крови и спинномозговой жидкости, а также молекулярно-генетическое тестирование биаллельных генов посредством секвенирования гена являются базовой диагностикой некетотической гиперглицинемии. Однако эффективного лечения тяжелой глициновой энцефалопатии на сегодняшний день не существует, а основная задача лечения заключается в снижении концентрации глицина бензоатом натрия в плазме путем блокирования глицинергических рецепторов на сайте рецептора N-метил-D-аспартата.
Конфликт интересов. Авторы заявляют об отсутствии какого-либо конфликта интересов при подготовке данной статьи.
Список литературы
1. Hennermann J.B. Prediction of long-term outcome in glycine encephalopathy: a clinical survey / J.B. Hennermann, J.M. Berger, U. Grieben et al. // Journal of Inherited Metabolic Disease. — 2012. — № 35(2). — P. 253-261.
2. Swanson M.A. Biochemical and molecular predictors for prognosis in nonketotic hyperglycinemia / M.A. Swanson, C.R. Coughlin, G.H. Scharer // Ann. Neurol. — 2015. — № 78(4). — P. 606-618.
3. Dinopoulos A. Atypical variants of nonketotic hyperglycinemia / A. Dinopoulos, Y. Matsubara, S. Kure // Mol. Genet. Metab. — 2005. — № 86(1–2). — P. 61-69.
4. Jiang T.J. Clinical and genetic analyses of a family with atypical nonketotic hyperglycinemia caused by compound heterozygous mutations in the GLDC gene / T.J. Jiang, J.J. Jiang, J.L. Xu // Zhongguo Dang Dai Er Ke Za Zhi. — 2017. — № 19. — P. 1087-1091.
5. Brunel-Guitton C. Late-onset nonketotic hyperglycinemia caused by a novel homozygous missense mutation in the GLDC gene / C. Brunel-Guitton, B. Casey, M. Coulter-Mackie // Mol. Genet. Metab. — 2011. — № 103(2). — P. 193-196.
6. Swanson M.A. Biochemical and molecular predictors for prognosis in nonketotic hyperglycinemia / M.A. Swanson, C.R. Jr Coughlin, G.H. Scharer et al. // Ann. Neurol. — 2015. — № 78(4). — P. 606-618.
7. Bjoraker K.J. Neurodevelopmental Outcome and Treatment Efficacy of Benzoate and Dextromethorphan in Siblings with Attenuated Nonketotic Hyperglycinemia // K.J. Bjoraker, M.A. Swanson, C.R. Coughlin et al. // J. Pediatr. — 2016. — № 170. — P. 234-239.
8. Ezgu F. Diagnosis of glycine encephalopathy in a pediatric patient by detection of a GLDC mutation during initial next generation DNA sequencing / F. Ezgu, B. Çiftci, B. Topçu et al. // Metab. Brain. Dis. — 2014. — № 29(1). — P. 211-213.
9. Chiu C.F. Nonketotic Hyperglycinemia of Infants in Taiwan / C.F. Chiu, J.L. Lin, J.J. Lin // Pediatr. Neonatol. — 2016. — № 57(5). — P. 420-426.
10. Tan E.S. Non-ketotic hyperglycinemia is usually not detec–table by tandem mass spectrometry newborn screening / E.S. Tan, V. Wiley, K. Carpenter et al. // Mol. Genet. Metab. — 2007. — № 90. — P. 446-448.
11. Applegarth D.A. Nonketotic hyperglycinemia (glycine ence–phalopathy): laboratory diagnosis / D.A. Applegarth, J.R. Toone // Mol. Genet. Metab. — 2001. — № 74. — P. 139-146.
12. Hoffmann G.F. Pyridoxal 5’-phosphate may be curative in early-onset epileptic encephalopathy / G.F. Hoffmann, B. Schmitt, M. Windfuhr et al. // J. Inherit. Metab. Dis. — 2007. — № 30. — P. 96-99.
13. Van Hove J. Mutation spectrum in non-ketotic hyperglycinemia. — Hamburg, Germany: Society for the Study of Inborn Errors of Metabolism Annual Symposium 2007. — Электронный ресурс: https://www.ncbi.nlm.nih.gov/books/NBK1357/.
14. Cusmai R. Ketogenic diet in early myoclonic encephalopathy due to non ketotic hyperglycinemia / R. Cusmai, D. Martinelli, R. Moavero et al. // Eur. J. Paediatr. Neurol. — 2012. — № 16. — P. 509-513.

/87-1.jpg)
/88-1.jpg)